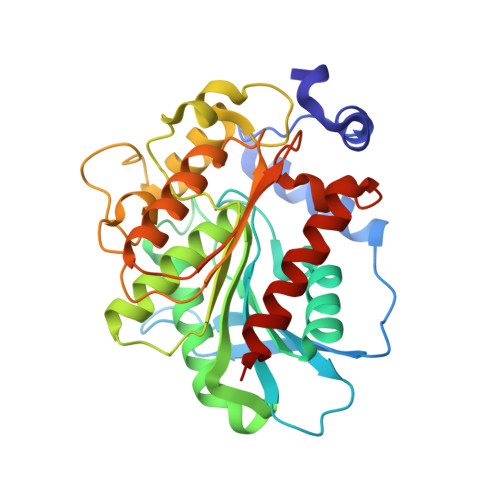
Structure and in silico simulations of a cold-active esterase reveals its prime cold-adaptation mechanism.
Noby, N., Auhim, H.S., Winter, S., Worthy, H.L., Embaby, A.M., Saeed, H., Hussein, A., Pudney, C.R., Rizkallah, P.J., Wells, S.A., Jones, D.D.(2021) Open Biol 11: 210182-210182
- PubMed: 34847772 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1098/rsob.210182
- Primary Citation Related Structures:
7B4Q - PubMed Abstract:
Here we determined the structure of a cold active family IV esterase (EstN7) cloned from Bacillus cohnii strain N1. EstN7 is a dimer with a classical α/β hydrolase fold. It has an acidic surface that is thought to play a role in cold-adaption by retaining solvation under changed water solvent entropy at lower temperatures. The conformation of the functionally important cap region is significantly different to EstN7's closest relatives, forming a bridge-like structure with reduced helical content providing greater access to the active site through more than one substrate access tunnel. However, dynamics do not appear to play a major role in cold adaption. Molecular dynamics at different temperatures, rigidity analysis, normal mode analysis and geometric simulations of motion confirm the flexibility of the cap region but suggest that the rest of the protein is largely rigid. Rigidity analysis indicates the distribution of hydrophobic tethers is appropriate to colder conditions, where the hydrophobic effect is weaker than in mesophilic conditions due to reduced water entropy. Thus, it is likely that increased substrate accessibility and tolerance to changes in water entropy are important for of EstN7's cold adaptation rather than changes in dynamics.
- Department of Biotechnology, Institute of Graduate Studies and Research, Alexandria University, Alexandria, Egypt.
Organizational Affiliation: