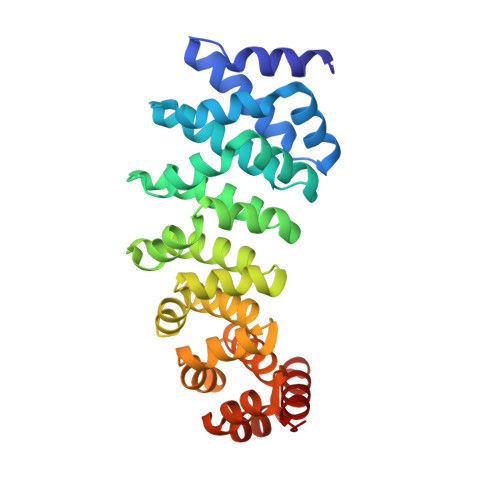
Structure-Guided Design of a Peptide Lock for Modular Peptide Binders.
Ernst, P., Zosel, F., Reichen, C., Nettels, D., Schuler, B., Pluckthun, A.(2020) ACS Chem Biol 15: 457-468
- PubMed: 31985201 Search on PubMed
- DOI: https://doi.org/10.1021/acschembio.9b00928
- Primary Citation Related Structures:
6S9L, 6S9M, 6S9N, 6S9O, 6S9P - PubMed Abstract:
Peptides play an important role in intermolecular interactions and are frequent analytes in diagnostic assays, also as unstructured, linear epitopes in whole proteins. Yet, due to the many different sequence possibilities even for short peptides, classical selection of binding proteins from a library, one at a time, is not scalable to proteomes. However, moving away from selection to a rational assembly of preselected modules binding to predefined linear epitopes would split the problem into smaller parts. These modules could then be reassembled in any desired order to bind to, in principle, arbitrary sequences, thereby circumventing any new rounds of selection. Designed Armadillo repeat proteins (dArmRPs) are modular, and they do bind elongated peptides in a modular way. Their consensus sequence carries pockets that prefer arginine and lysine. In our quest to select pockets for all amino acid side chains, we had discovered that repetitive sequences can lead to register shifts and peptide flipping during selections from libraries, hindering the selection of new binding specificities. To solve this problem, we now created an orthogonal binding specificity by a combination of grafting from β-catenin, computational design and mutual optimization of the pocket and the bound peptide. We have confirmed the design and the desired interactions by X-ray structure determination. Furthermore, we could confirm the absence of sliding in solution by a single-molecule Förster resonance energy transfer. The new pocket could be moved from the N-terminus of the protein to the middle, retaining its properties, further underlining the modularity of the system.
- Department of Biochemistry , University Zürich , Winterthurerstrasse 190 , 8057 Zürich , Switzerland.
Organizational Affiliation: