Re-evaluation of low-resolution crystal structures via interactive molecular-dynamics flexible fitting (iMDFF): a case study in complement C4.
Croll, T.I., Andersen, G.R.(2016) Acta Crystallogr D Struct Biol 72: 1006-1016
- PubMed: 27599733 Search on PubMed
- DOI: https://doi.org/10.1107/S2059798316012201
- Primary Citation Related Structures:
5JPM, 5JPN, 5JTW - PubMed Abstract:
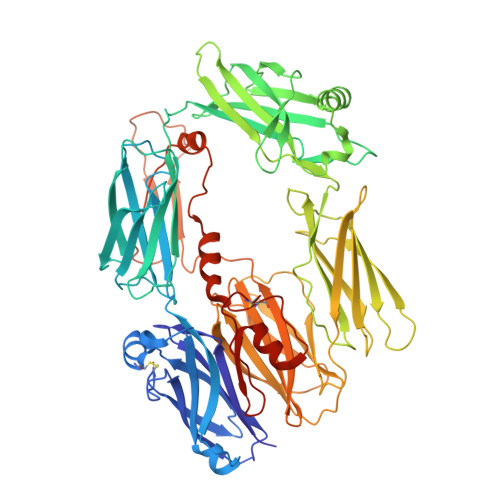
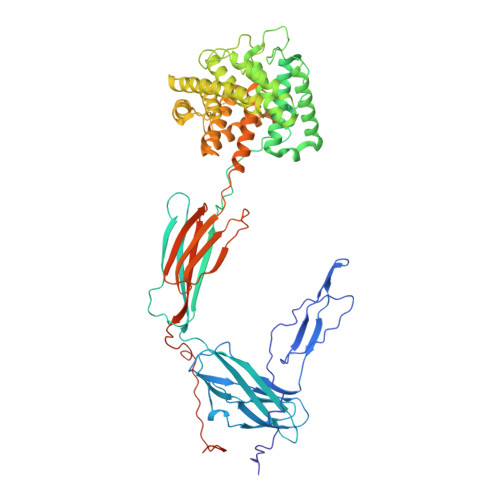
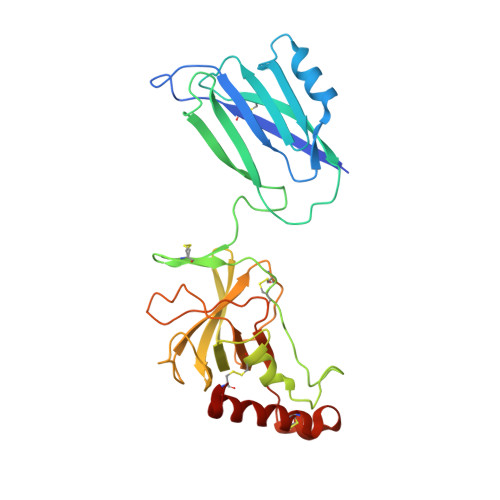
While the rapid proliferation of high-resolution structures in the Protein Data Bank provides a rich set of templates for starting models, it remains the case that a great many structures both past and present are built at least in part by hand-threading through low-resolution and/or weak electron density. With current model-building tools this task can be challenging, and the de facto standard for acceptable error rates (in the form of atomic clashes and unfavourable backbone and side-chain conformations) in structures based on data with dmax not exceeding 3.5 Å reflects this. When combined with other factors such as model bias, these residual errors can conspire to make more serious errors in the protein fold difficult or impossible to detect. The three recently published 3.6-4.2 Å resolution structures of complement C4 (PDB entries 4fxg, 4fxk and 4xam) rank in the top quartile of structures of comparable resolution both in terms of Rfree and MolProbity score, yet, as shown here, contain register errors in six β-strands. By applying a molecular-dynamics force field that explicitly models interatomic forces and hence excludes most physically impossible conformations, the recently developed interactive molecular-dynamics flexible fitting (iMDFF) approach significantly reduces the complexity of the conformational space to be searched during manual rebuilding. This substantially improves the rate of detection and correction of register errors, and allows user-guided model building in maps with a resolution lower than 3.5 Å to converge to solutions with a stereochemical quality comparable to atomic resolution structures. Here, iMDFF has been used to individually correct and re-refine these three structures to MolProbity scores of <1.7, and strategies for working with such challenging data sets are suggested. Notably, the improved model allowed the resolution for complement C4b to be extended from 4.2 to 3.5 Å as demonstrated by paired refinement.
- Institute of Health and Biomedical Innovation, Queensland University of Technology, GPO Box 2434, Brisbane, QLD 4001, Australia.
Organizational Affiliation: