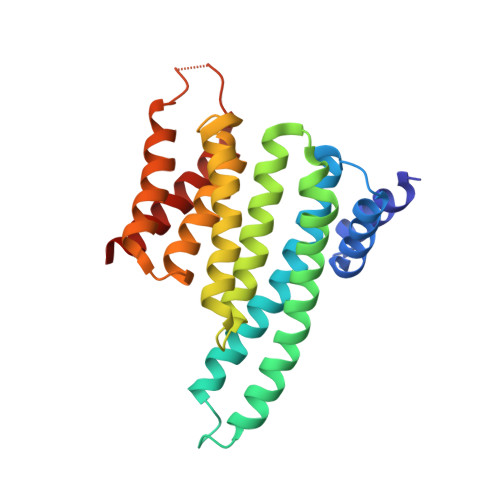
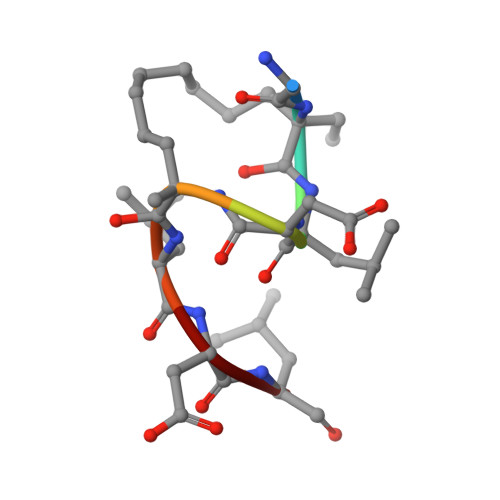
Adapting free energy perturbation simulations for large macrocyclic ligands: how to dissect contributions from direct binding and free ligand flexibility.
Wallraven, K., Holmelin, F.L., Glas, A., Hennig, S., Frolov, A.I., Grossmann, T.N.(2020) Chem Sci 11: 2269-2276
- PubMed: 32180932 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1039/c9sc04705k
- Primary Citation Related Structures:
6RLZ - PubMed Abstract:
Large and flexible ligands gain increasing interest in the development of bioactive agents. They challenge the applicability of computational ligand optimization strategies originally developed for small molecules. Free energy perturbation (FEP) is often used for predicting binding affinities of small molecule ligands, however, its use for more complex ligands remains limited. Herein, we report the structure-based design of peptide macrocycles targeting the protein binding site of human adaptor protein 14-3-3. We observe a surprisingly strong dependency of binding affinities on relatively small variations in substituent size. FEP was performed to rationalize observed trends. To account for insufficient convergence of FEP, restrained calculations were performed and complemented with extensive REST MD simulations of the free ligands. These calculations revealed that changes in affinity originate both from altered direct interactions and conformational changes of the free ligand. In addition, MD simulations provided the basis to rationalize unexpected trends in ligand lipophilicity. We also verified the anticipated interaction site and binding mode for one of the high affinity ligands by X-ray crystallography. The introduced fully-atomistic simulation protocol can be used to rationalize the development of structurally complex ligands which will support future ligand maturation efforts.
- Department of Chemistry & Pharmaceutical Sciences , VU University Amsterdam , De Boelelaan 1083 , 1081 HV Amsterdam , The Netherlands . Email: t.n.grossmann@vu.nl.
Organizational Affiliation: