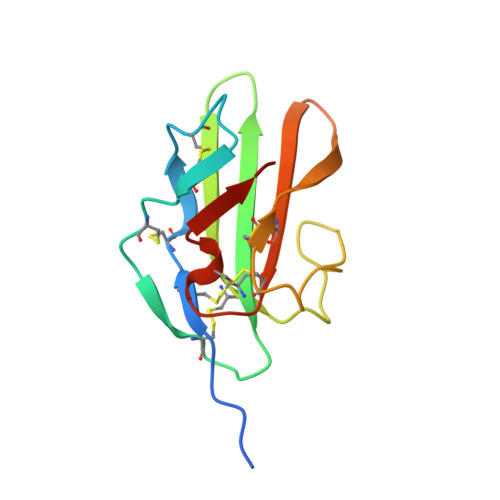
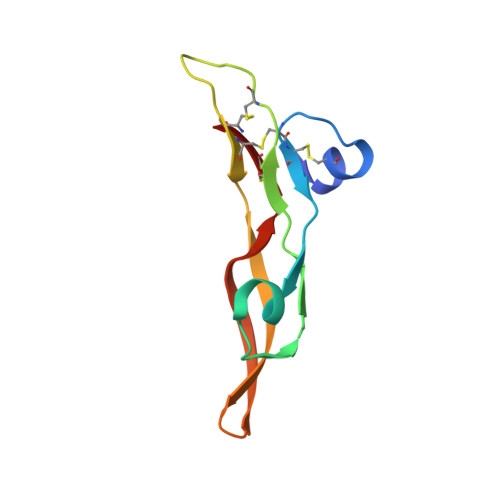
An engineered transforming growth factor beta (TGF-beta ) monomer that functions as a dominant negative to block TGF-beta signaling.
Kim, S.K., Barron, L., Hinck, C.S., Petrunak, E.M., Cano, K.E., Thangirala, A., Iskra, B., Brothers, M., Vonberg, M., Leal, B., Richter, B., Kodali, R., Taylor, A.B., Du, S., Barnes, C.O., Sulea, T., Calero, G., Hart, P.J., Hart, M.J., Demeler, B., Hinck, A.P.(2017) J Biol Chem 292: 7173-7188
- PubMed: 28228478 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1074/jbc.M116.768754
- Primary Citation Related Structures:
5TX2, 5TX4, 5TX6 - PubMed Abstract:
The transforming growth factor β isoforms, TGF-β1, -β2, and -β3, are small secreted homodimeric signaling proteins with essential roles in regulating the adaptive immune system and maintaining the extracellular matrix. However, dysregulation of the TGF-β pathway is responsible for promoting the progression of several human diseases, including cancer and fibrosis. Despite the known importance of TGF-βs in promoting disease progression, no inhibitors have been approved for use in humans. Herein, we describe an engineered TGF-β monomer, lacking the heel helix, a structural motif essential for binding the TGF-β type I receptor (TβRI) but dispensable for binding the other receptor required for TGF-β signaling, the TGF-β type II receptor (TβRII), as an alternative therapeutic modality for blocking TGF-β signaling in humans. As shown through binding studies and crystallography, the engineered monomer retained the same overall structure of native TGF-β monomers and bound TβRII in an identical manner. Cell-based luciferase assays showed that the engineered monomer functioned as a dominant negative to inhibit TGF-β signaling with a K i of 20-70 nm Investigation of the mechanism showed that the high affinity of the engineered monomer for TβRII, coupled with its reduced ability to non-covalently dimerize and its inability to bind and recruit TβRI, enabled it to bind endogenous TβRII but prevented it from binding and recruiting TβRI to form a signaling complex. Such engineered monomers provide a new avenue to probe and manipulate TGF-β signaling and may inform similar modifications of other TGF-β family members.
- the Departments of Biochemistry and Structural Biology and.
Organizational Affiliation: