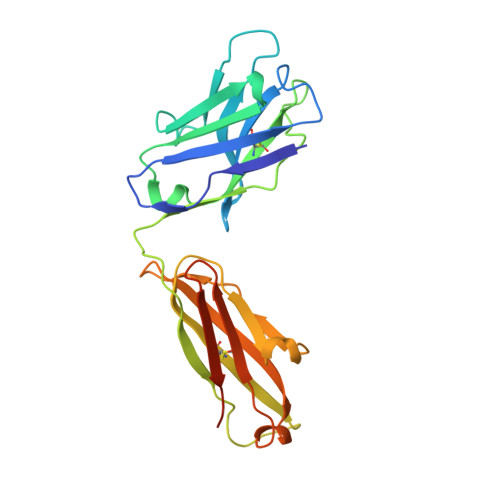
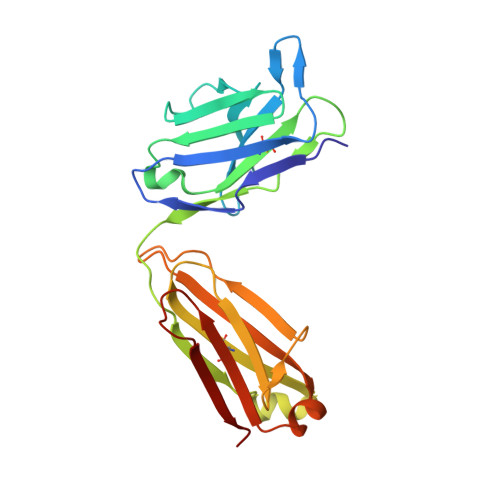
Principles for computational design of binding antibodies.
Baran, D., Pszolla, M.G., Lapidoth, G.D., Norn, C., Dym, O., Unger, T., Albeck, S., Tyka, M.D., Fleishman, S.J.(2017) Proc Natl Acad Sci U S A 114: 10900-10905
- PubMed: 28973872 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.1707171114
- Primary Citation Related Structures:
5NB5, 5NBI - PubMed Abstract:
Natural proteins must both fold into a stable conformation and exert their molecular function. To date, computational design has successfully produced stable and atomically accurate proteins by using so-called "ideal" folds rich in regular secondary structures and almost devoid of loops and destabilizing elements, such as cavities. Molecular function, such as binding and catalysis, however, often demands nonideal features, including large and irregular loops and buried polar interaction networks, which have remained challenging for fold design. Through five design/experiment cycles, we learned principles for designing stable and functional antibody variable fragments (Fvs). Specifically, we ( i ) used sequence-design constraints derived from antibody multiple-sequence alignments, and ( ii ) during backbone design, maintained stabilizing interactions observed in natural antibodies between the framework and loops of complementarity-determining regions (CDRs) 1 and 2. Designed Fvs bound their ligands with midnanomolar affinities and were as stable as natural antibodies, despite having >30 mutations from mammalian antibody germlines. Furthermore, crystallographic analysis demonstrated atomic accuracy throughout the framework and in four of six CDRs in one design and atomic accuracy in the entire Fv in another. The principles we learned are general, and can be implemented to design other nonideal folds, generating stable, specific, and precise antibodies and enzymes.
- Department of Biomolecular Sciences, Weizmann Institute of Science, Rehovot 76100, Israel.
Organizational Affiliation: