Replace: A Strategy for Iterative Design of Cyclin- Binding Groove Inhibitors
Andrews, M.J., Kontopidis, G., Mcinnes, C., Plater, A., Innes, L., Cowan, A., Jewsbury, P., Fischer, P.M.(2006) Chembiochem 7: 1909
- PubMed: 17051658 Search on PubMed
- DOI: https://doi.org/10.1002/cbic.200600189
- Primary Citation Related Structures:
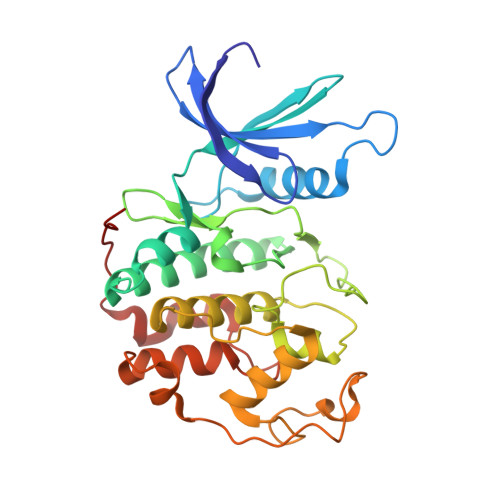
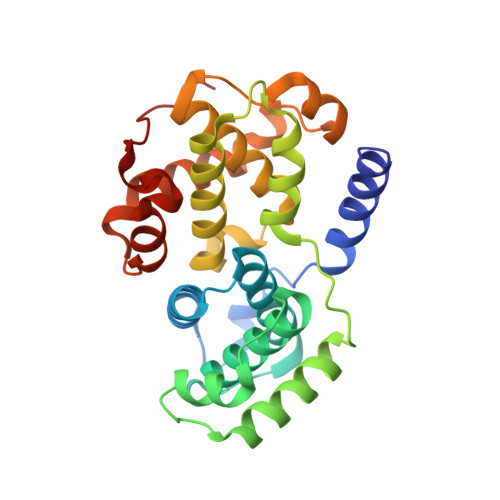
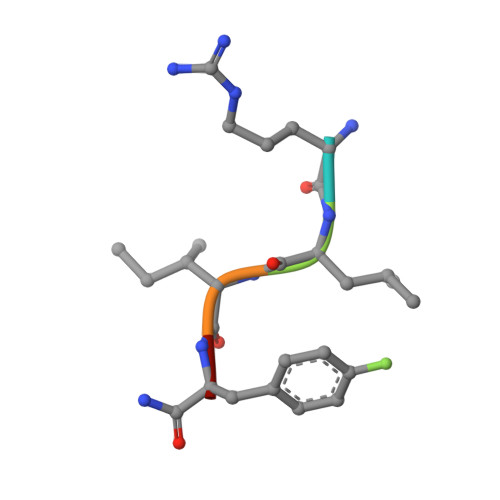
2UUE, 2V22 - PubMed Abstract:
We describe a drug-design strategy termed REPLACE (REplacement with Partial Ligand Alternatives through Computational Enrichment) in which nonpeptidic surrogates for specific determinants of known peptide ligands are identified in silico by using a core peptide-bound protein structure as a design anchor. In the REPLACE application example, we present the effective replacement of two critical binding motifs in a lead protein-protein interaction inhibitor pentapeptide with more druglike phenyltriazole and diphenyl ether groups. These were identified through docking of fragment libraries into the volume of the cyclin-binding groove of CDK2/cyclin A vacated through truncation of the inhibitor peptide-binding determinants. Proof of concept for this strategy was obtained through the generation of potent peptide-small-molecule hybrids and by the confirmation of inhibitor-binding modes in X-ray crystal structures. This method therefore allows nonpeptide fragments to be identified without the requirement for a high-sensitivity binding assay and should be generally applicable in replacing amino acids as individual residues or groups in peptide inhibitors to generate pharmaceutically acceptable lead molecules.
- Cyclacel Limited, James Lindsay Place, Dundee, DD1 5JJ, UK. martin.andrews@glpg.com
Organizational Affiliation: