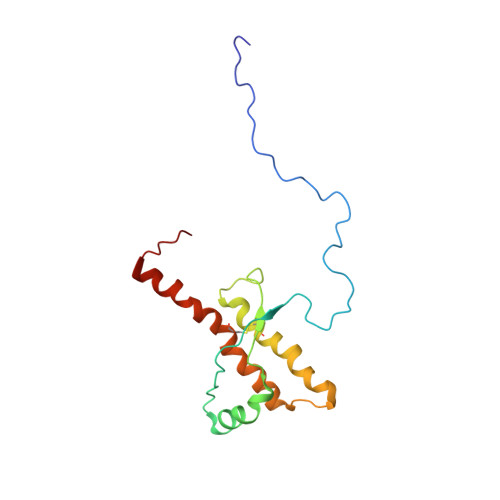
NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features.
Ilc, G., Giachin, G., Jaremko, M., Jaremko, L., Benetti, F., Plavec, J., Zhukov, I., Legname, G.(2010) PLoS One 5: e11715-e11715
- PubMed: 20661422 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1371/journal.pone.0011715
- Primary Citation Related Structures:
2KUN - PubMed Abstract:
Prion diseases are fatal neurodegenerative disorders caused by an aberrant accumulation of the misfolded cellular prion protein (PrP(C)) conformer, denoted as infectious scrapie isoform or PrP(Sc). In inherited human prion diseases, mutations in the open reading frame of the PrP gene (PRNP) are hypothesized to favor spontaneous generation of PrP(Sc) in specific brain regions leading to neuronal cell degeneration and death. Here, we describe the NMR solution structure of the truncated recombinant human PrP from residue 90 to 231 carrying the Q212P mutation, which is believed to cause Gerstmann-Sträussler-Scheinker (GSS) syndrome, a familial prion disease. The secondary structure of the Q212P mutant consists of a flexible disordered tail (residues 90-124) and a globular domain (residues 125-231). The substitution of a glutamine by a proline at the position 212 introduces novel structural differences in comparison to the known wild-type PrP structures. The most remarkable differences involve the C-terminal end of the protein and the beta(2)-alpha(2) loop region. This structure might provide new insights into the early events of conformational transition of PrP(C) into PrP(Sc). Indeed, the spontaneous formation of prions in familial cases might be due to the disruptions of the hydrophobic core consisting of beta(2)-alpha(2) loop and alpha(3) helix.
- Slovenian NMR Centre, National Institute of Chemistry, Ljubljana, Slovenia.
Organizational Affiliation: