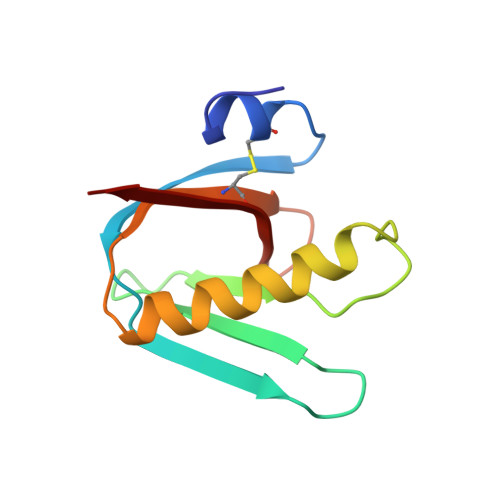
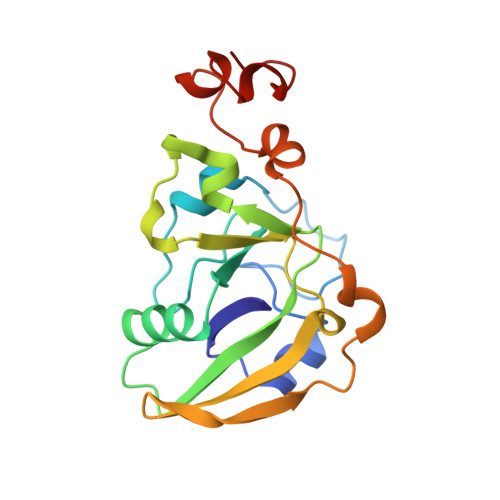
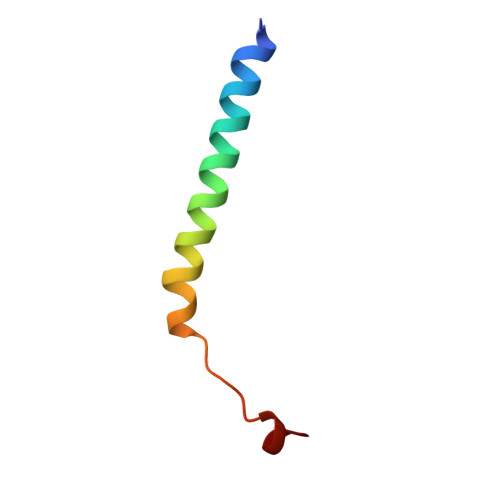
Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin.
Sixma, T.K., Van Zanten, B.A.M., Dauter, Z., Hol, W.G.J.(1993) J Mol Biology 230: 890-918
- PubMed: 8478941 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.1993.1209
- Primary Citation Related Structures:
1LTS - PubMed Abstract:
Heat-labile enterotoxin (LT) from Escherichia coli is a bacterial protein toxin with an AB5 multimer structure, in which the B pentamer has a membrane binding function and the A subunit is needed for enzymatic activity. The LT crystal structure has been solved using a combination of multiple isomorphous replacement, fivefold averaging and molecular dynamics refinement. Phase combination using all these sources of phase information was of crucial importance for the chain tracing. The structure has now been refined to 1.95 A resolution, resulting in a model containing 6035 protein atoms and 293 solvent molecules with a crystallographic R-factor of 18.2% and good stereochemistry. The B subunits are arranged as a highly stable pentamer with a donut shape. Each subunit takes part in approximately 30 inter-subunit hydrogen bonds and six salt bridges with its two neighbors, whilst burying a large surface area. The A subunit has higher temperature factors and less well-defined secondary structure than the B subunits. It interacts with the B pentamer mainly via the C-terminal A2 fragment, which runs through the highly charged central pore of the B subunits. The pore contains at least 66 water molecules, which fill the space left by the A2 fragment. A detailed analysis of the contacts between A and B subunits showed that most specific contacts occur at the entrance of the central pore of the B pentamer, while the contacts within the pore are mainly hydrophobic and water mediated, with the exception of two salt bridges. Only a few contacts exist between the A1 fragment and the B pentamer, showing that the A2 fragment functions as a "linker" of the A and B parts of the protein. Interacting with the A subunit by the B subunits does not cause large deviations from a common B subunit structure, and the 5-fold symmetry is well maintained. A potential NAD(+)-binding site is located in an elongated crevice at the interface of two small sheets in the A1 fragment. At the back of this crevice the functionally important Arg7 makes a hydrogen bond connecting two strands, which seems to be conserved across the ADP-ribosylating toxin family. The putative catalytic residue (A1:Glu112) is located nearby, close to a very hydrophobic region, which packs two loops together. This hydrophobic region may be important for catalysis and membrane translocation.
- BIOSON Research Institute, University of Groningen, The Netherlands.
Organizational Affiliation: