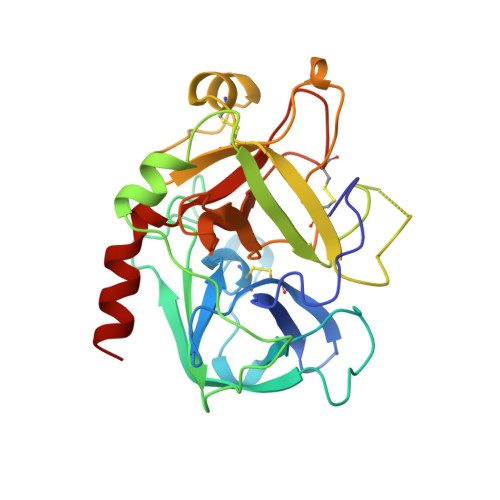
The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships.
Bode, W., Turk, D., Karshikov, A.(1992) Protein Sci 1: 426-471
- PubMed: 1304349 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1002/pro.5560010402
- Primary Citation Related Structures:
1AI8, 1AIX - PubMed Abstract:
Thrombin is a multifunctional serine proteinase that plays a key role in coagulation while exhibiting several other key cellular bioregulatory functions. The X-ray crystal structure of human alpha-thrombin was determined in its complex with the specific thrombin inhibitor D-Phe-Pro-Arg chloromethylketone (PPACK) using Patterson search methods and a search model derived from trypsinlike proteinases of known spatial structure (Bode, W., Mayr, I., Baumann, U., Huber, R., Stone, S.R., & Hofsteenge, J., 1989, EMBO J. 8, 3467-3475). The crystallographic refinement of the PPACK-thrombin model has now been completed at an R value of 0.156 (8 to 1.92 A); in particular, the amino- and the carboxy-termini of the thrombin A-chain are now defined and all side-chain atoms localized; only proline 37 was found to be in a cis-peptidyl conformation. The thrombin B-chain exhibits the characteristic polypeptide fold of trypsinlike serine proteinases; 195 residues occupy topologically equivalent positions with residues in bovine trypsin and 190 with those in bovine chymotrypsin with a root-mean-square (r.m.s.) deviation of 0.8 A for their alpha-carbon atoms. Most of the inserted residues constitute novel surface loops. A chymotrypsinogen numbering is suggested for thrombin based on the topological equivalences. The thrombin A-chain is arranged in a boomeranglike shape against the B-chain globule opposite to the active site; it resembles somewhat the propeptide of chymotrypsin(ogen) and is similarly not involved in substrate and inhibitor binding. Thrombin possesses an exceptionally large proportion of charged residues. The negatively and positively charged residues are not distributed uniformly over the whole molecule, but are clustered to form a sandwichlike electrostatic potential; in particular, two extended patches of mainly positively charged residues occur close to the carboxy-terminal B-chain helix (forming the presumed heparin-binding site) and on the surface of loop segment 70-80 (the fibrin[ogen] secondary binding exosite), respectively; the negatively charged residues are more clustered in the ringlike region between both poles, particularly around the active site. Several of the charged residues are involved in salt bridges; most are on the surface, but 10 charged protein groups form completely buried salt bridges and clusters. These electrostatic interactions play a particularly important role in the intrachain stabilization of the A-chain, in the coherence between the A- and the B-chain, and in the surface structure of the fibrin(ogen) secondary binding exosite (loop segment 67-80).(ABSTRACT TRUNCATED AT 400 WORDS)
- Max-Planck-Institut für Biochemie, Martinsried, Germany.
Organizational Affiliation: