ADAPT-M: A workflow for rapid, quantitative in vitro measurements of enriched protein libraries.
Perez, C.P., DelRosso, N.V., Noland, C.L., Parekh, U., Choe, C.A., Eguchi, R.R., Wen, Q., Fordyce, P.M., Huang, P.S.(2025) bioRxiv
- PubMed: 41279512 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1101/2025.10.21.683815
- Primary Citation Related Structures:
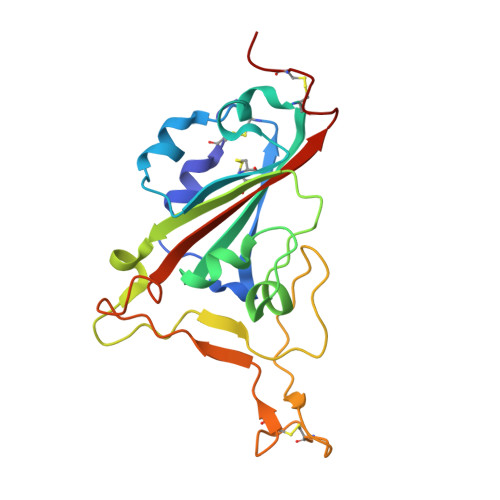
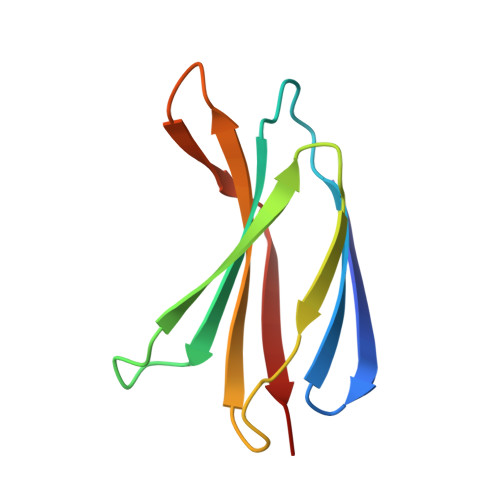
9Y5Y - PubMed Abstract:
Protein-protein interactions underpin most cellular interactions, and engineered binders present powerful tools for probing biology and developing novel therapeutics. One bottleneck in binder generation is the scalable, quantitative characterization of these interactions. We present ADAPT-M ( A ffinity D etermination by A daptation of P ro T ein binders for M icrofluidics), a streamlined workflow that connects yeast surface display (YSD) with in vitro affinity and kinetic measurements using the high-throughput STAMMPPING microfluidic platform. ADAPT-M quantifies K d s and dissociation kinetic parameters for hundreds of enriched protein variants in under one week without requiring hands-on protein purification. We applied ADAPT-M to a computationally designed library targeting the SARS-CoV-2 Omicron BA.1 receptor binding domain, successfully recovering and measuring K d s for most highly enriched YSD variants. Measurements correlate strongly with biolayer interferometry and yeast titration assays. ADAPT-M further enabled selection of lead candidates for structural and mutational analysis, which revealed designed paratopes were preserved despite binding to off-target epitopes. By bridging YSD screening and in vitro validation, ADAPT-M accelerates protein binder discovery and supports data-driven protein engineering.
- Biophysics Program, Stanford University, Stanford, CA, USA.
Organizational Affiliation: