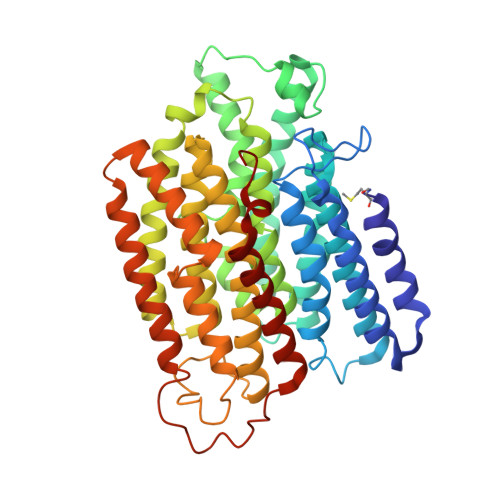
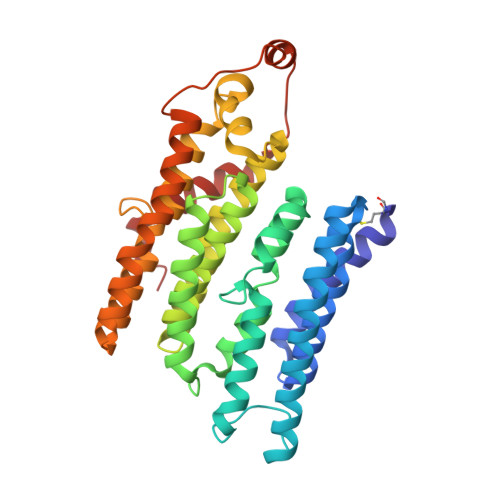
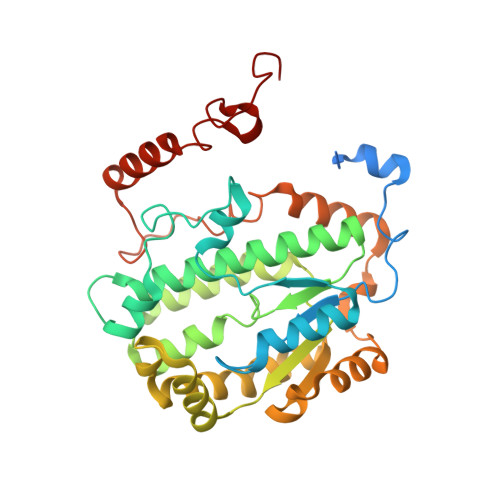
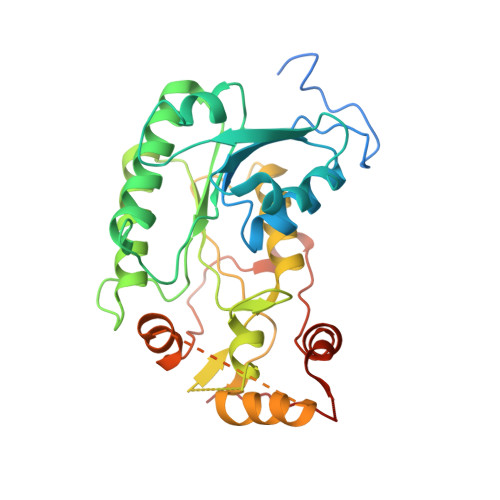
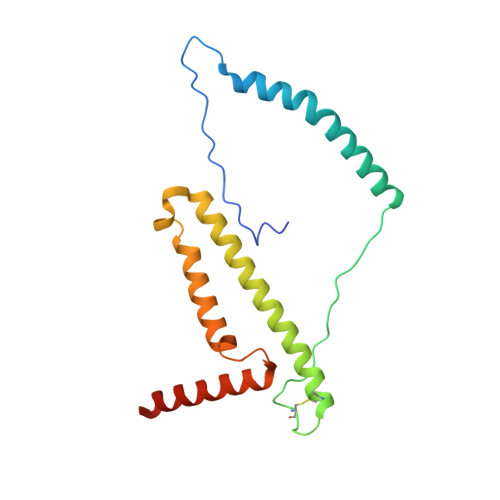
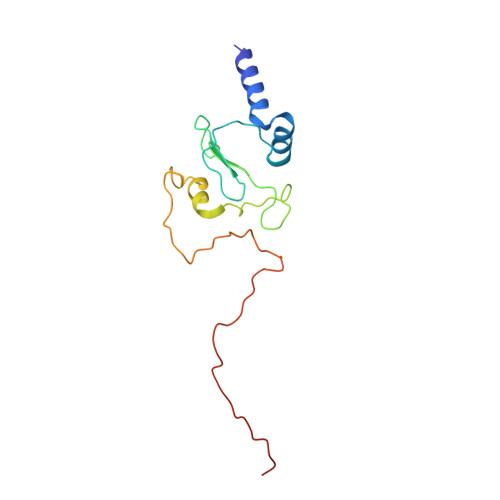
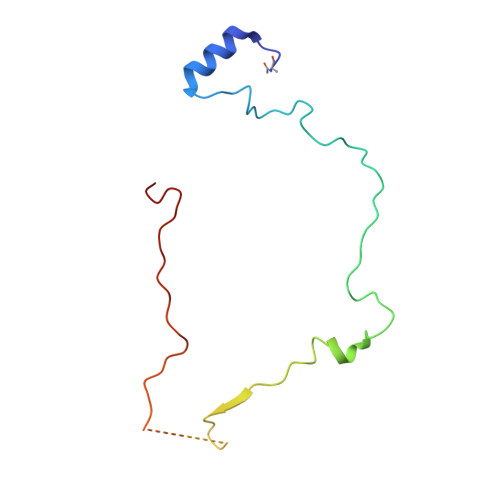
Structural basis for a complex I mutation that blocks pathological ROS production.
Yin, Z., Burger, N., Kula-Alwar, D., Aksentijevic, D., Bridges, H.R., Prag, H.A., Grba, D.N., Viscomi, C., James, A.M., Mottahedin, A., Krieg, T., Murphy, M.P., Hirst, J.(2021) Nat Commun 12: 707-707
- PubMed: 33514727 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/s41467-021-20942-w
- Primary Citation Related Structures:
7AK5, 7AK6 - PubMed Abstract:
Mitochondrial complex I is central to the pathological reactive oxygen species (ROS) production that underlies cardiac ischemia-reperfusion (IR) injury. ND6-P25L mice are homoplasmic for a disease-causing mtDNA point mutation encoding the P25L substitution in the ND6 subunit of complex I. The cryo-EM structure of ND6-P25L complex I revealed subtle structural changes that facilitate rapid conversion to the "deactive" state, usually formed only after prolonged inactivity. Despite its tendency to adopt the "deactive" state, the mutant complex is fully active for NADH oxidation, but cannot generate ROS by reverse electron transfer (RET). ND6-P25L mitochondria function normally, except for their lack of RET ROS production, and ND6-P25L mice are protected against cardiac IR injury in vivo. Thus, this single point mutation in complex I, which does not affect oxidative phosphorylation but renders the complex unable to catalyse RET, demonstrates the pathological role of ROS production by RET during IR injury.
- MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Organizational Affiliation: