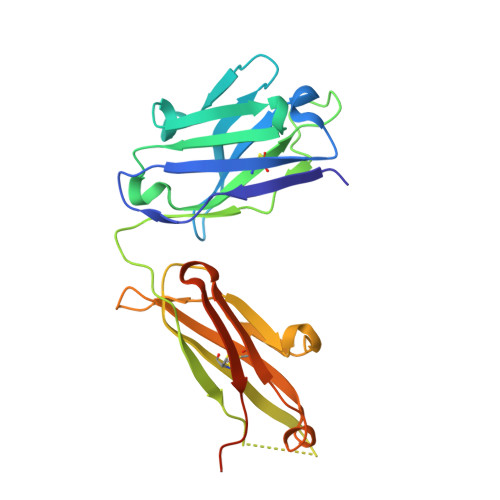
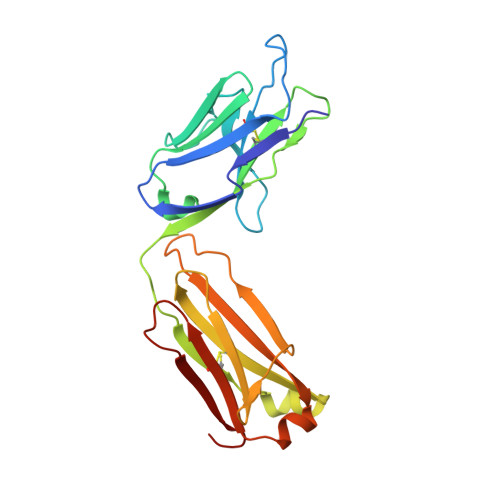
Binding symmetry and surface flexibility mediate antibody self-association.
Schrag, J.D., Picard, M.E., Gaudreault, F., Gagnon, L.P., Baardsnes, J., Manenda, M.S., Sheff, J., Deprez, C., Baptista, C., Hogues, H., Kelly, J.F., Purisima, E.O., Shi, R., Sulea, T.(2019) MAbs 11: 1300-1318
- PubMed: 31318308 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1080/19420862.2019.1632114
- Primary Citation Related Structures:
6MXR, 6MXS, 6MY4, 6MY5 - PubMed Abstract:
Solution stability is an important factor in the optimization of engineered biotherapeutic candidates such as monoclonal antibodies because of its possible effects on manufacturability, pharmacology, efficacy and safety. A detailed atomic understanding of the mechanisms governing self-association of natively folded protein monomers is required to devise predictive tools to guide screening and re-engineering along the drug development pipeline. We investigated pairs of affinity-matured full-size antibodies and observed drastically different propensities to aggregate from variants differing by a single amino-acid. Biophysical testing showed that antigen-binding fragments (Fabs) from the aggregating antibodies also reversibly associated with equilibrium dissociation constants in the low-micromolar range. Crystal structures (PDB accession codes 6MXR, 6MXS, 6MY4, 6MY5) and bottom-up hydrogen-exchange mass spectrometry revealed that Fab self-association occurs in a symmetric mode that involves the antigen complementarity-determining regions. Subtle local conformational changes incurred upon point mutation of monomeric variants foster formation of complementary polar interactions and hydrophobic contacts to generate a dimeric Fab interface. Testing of popular in silico tools generally indicated low reliabilities for predicting the aggregation propensities observed. A structure-aggregation data set is provided here in order to stimulate further improvements of in silico tools for prediction of native aggregation. Incorporation of intermolecular docking, conformational flexibility, and short-range packing interactions may all be necessary features of the ideal algorithm.
- Human Health Therapeutics Research Centre, National Research Council Canada , Montreal , QC H4P 2R2 , Canada.
Organizational Affiliation: