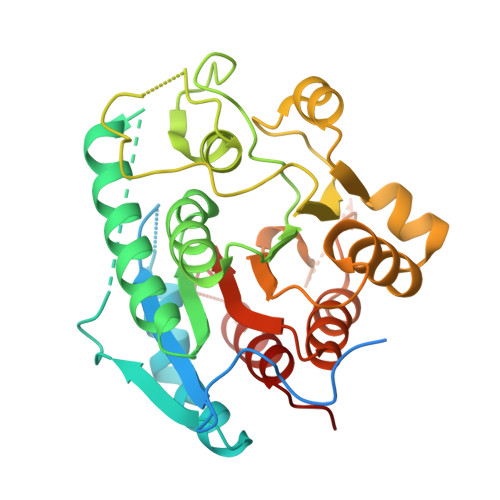
Crystal structure of a cold-active arginase from Glaciozyma antarctica.
Quay, D.H.X., Yusof, N.Y., Jonet, M.A., Kamaruddin, S., Md Illias, R., Mahadi, N.M., Keep, N., Firdaus-Raih, M., Abu Bakar, F.D., Abdul Murad, A.M.(2026) Acta Crystallogr F Struct Biol Commun 82: 167-175
- PubMed: 41973519 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1107/S2053230X26003092
- Primary Citation Related Structures:
5YNL - PubMed Abstract:
Arginase is a metal-dependent metalloenzyme that catalyses the hydrolysis of L-arginine to L-ornithine and urea and is widely distributed across animals, bacteria, fungi and protozoa. Here, we report the first three-dimensional crystal structure of a cold-active arginase from the psychrophilic yeast Glaciozyma antarctica (GaArg). The apo structure was solved at 2.35 Å resolution in space group H3 by molecular replacement using an AlphaFold-generated model. GaArg adopts a conserved αβα sandwich fold similar to previously characterized arginases. The crystal structure reveals four regions lacking interpretable electron density, three of which are located near the entrance to the active site. Despite conservation of the identities and the coordination geometry of metal-binding residues, the apo GaArg structure exhibits broadly conserved ligand-binding residue orientations relative to homologous arginases, with minor conformational differences and a disordered loop corresponding to a substrate-interacting region. Analytical size-exclusion chromatography and multimer prediction support the hexameric assembly of GaArg in solution. Quantitative analysis of intramolecular interactions indicates that GaArg contains fewer hydrogen bonds than mesophilic and thermophilic homologues, while its salt-bridge content is comparable to that of the mesophilic enzyme but lower than that of the thermophilic homologue. These features are consistent with a modest reduction in structural rigidity associated with cold adaptation. Ligand- and metal-bound structures will be required to establish their contributions to cold-adaptation.
- Department of Applied Physics, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 Bangi, Selangor, Malaysia.
Organizational Affiliation: