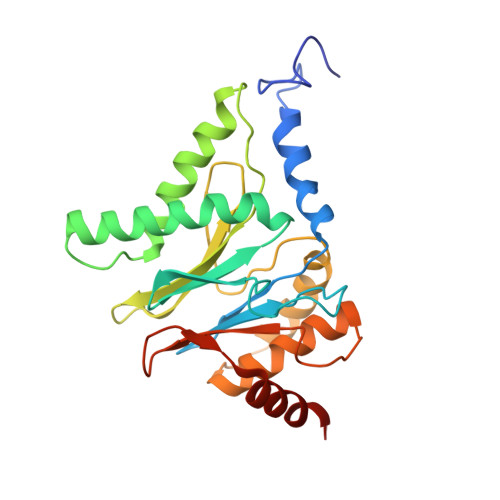
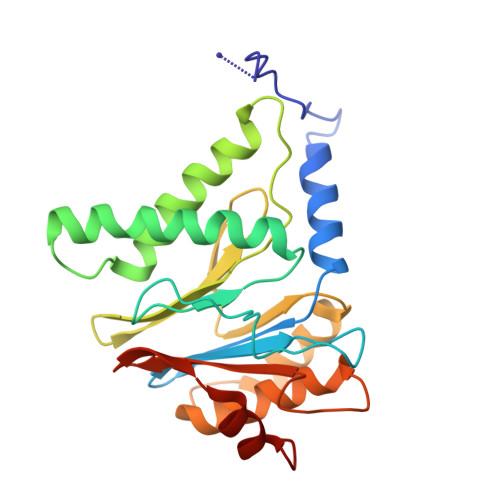
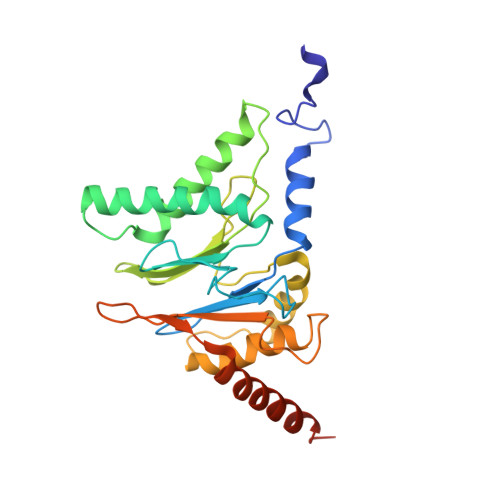
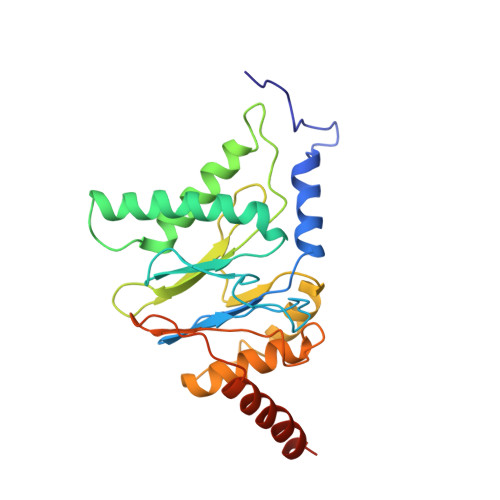
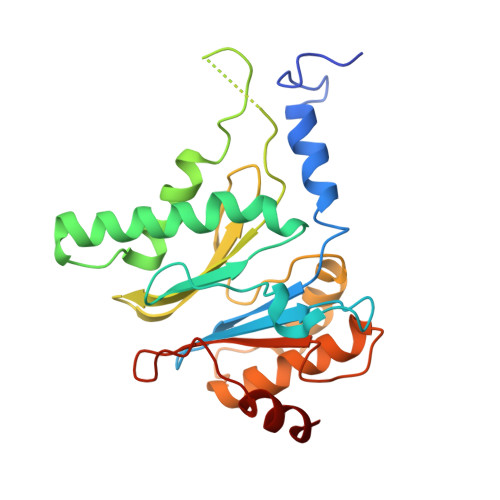
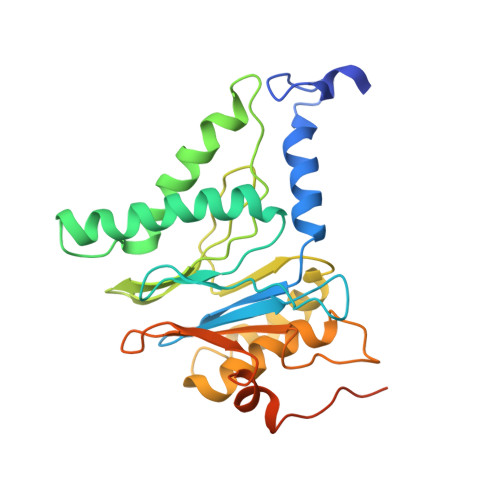
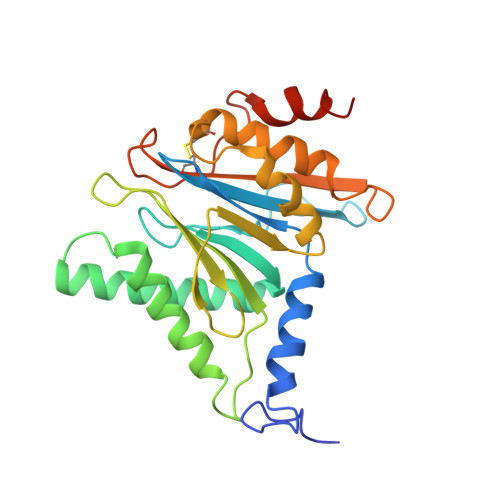
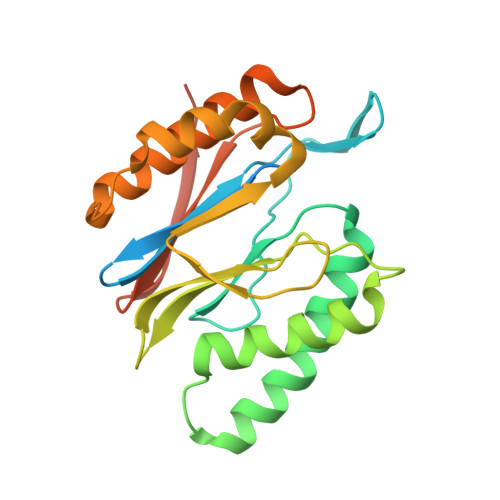
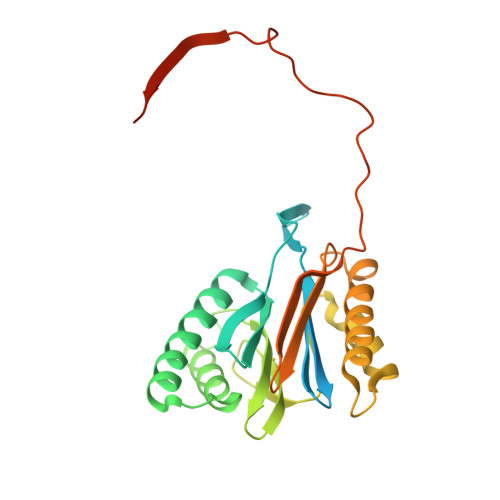
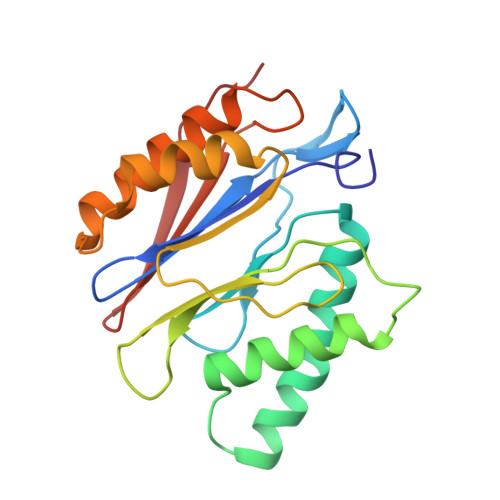
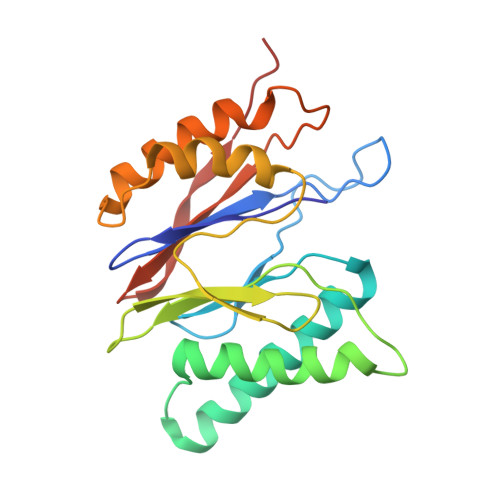
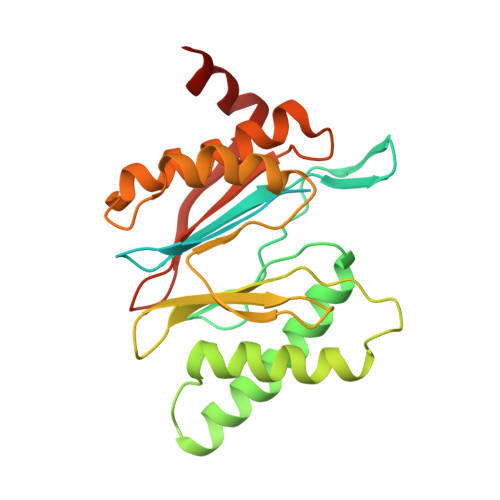
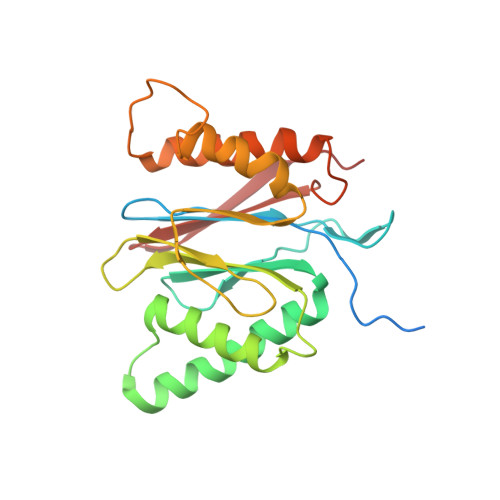
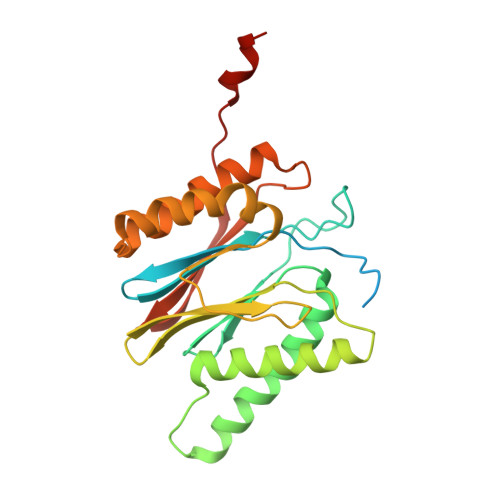
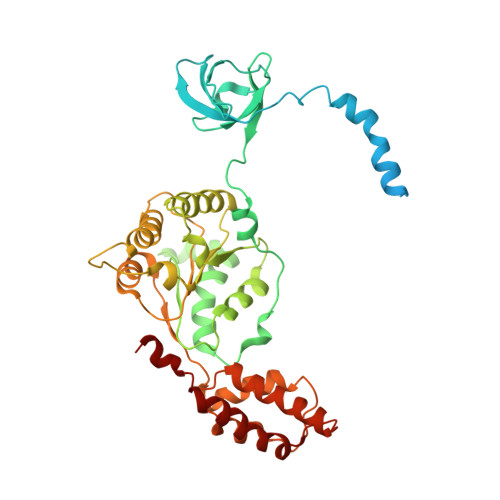
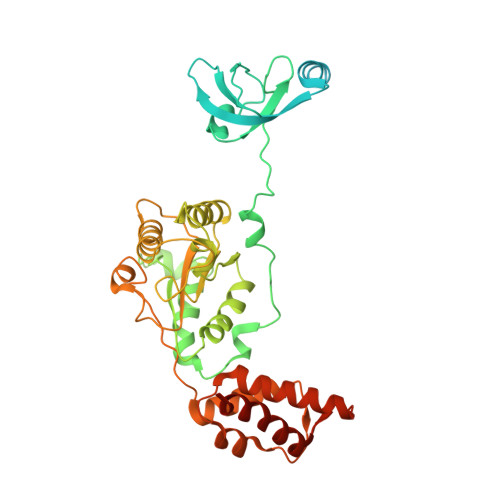
Structural basis for dynamic regulation of the human 26S proteasome.
Chen, S., Wu, J., Lu, Y., Ma, Y.B., Lee, B.H., Yu, Z., Ouyang, Q., Finley, D.J., Kirschner, M.W., Mao, Y.(2016) Proc Natl Acad Sci U S A 113: 12991-12996
- PubMed: 27791164 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.1614614113
- Primary Citation Related Structures:
5T0C, 5T0G, 5T0H, 5T0I, 5T0J - PubMed Abstract:
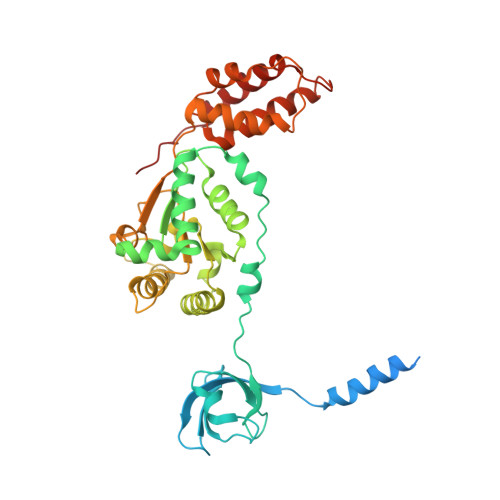
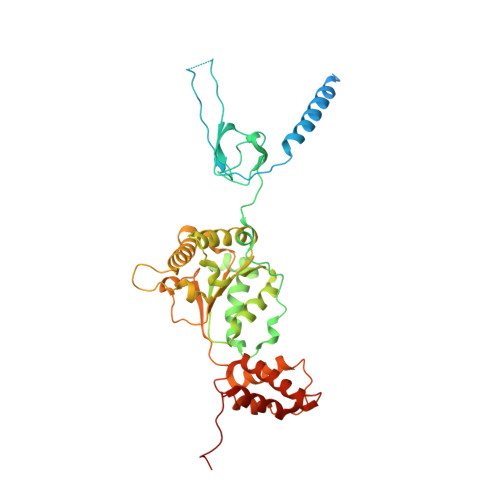
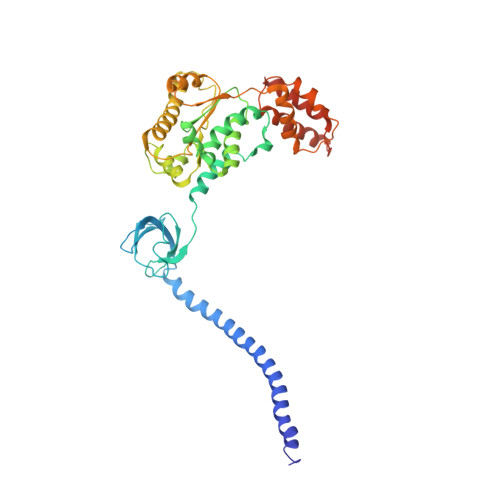
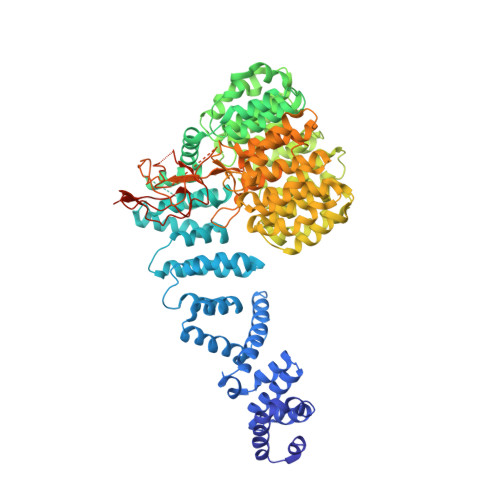
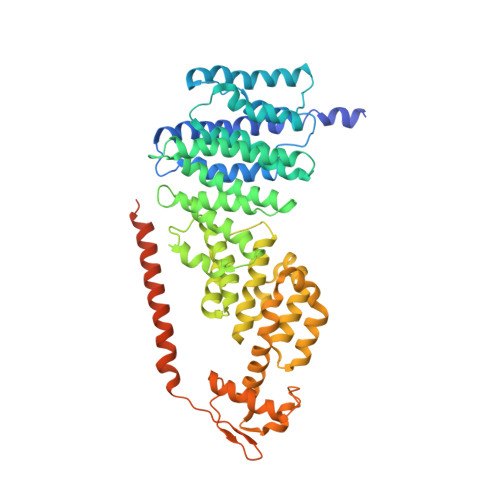
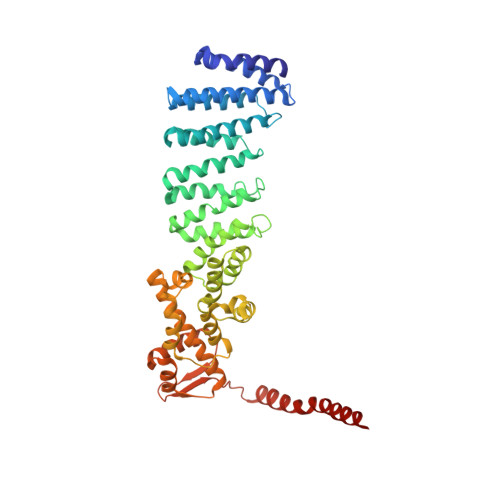
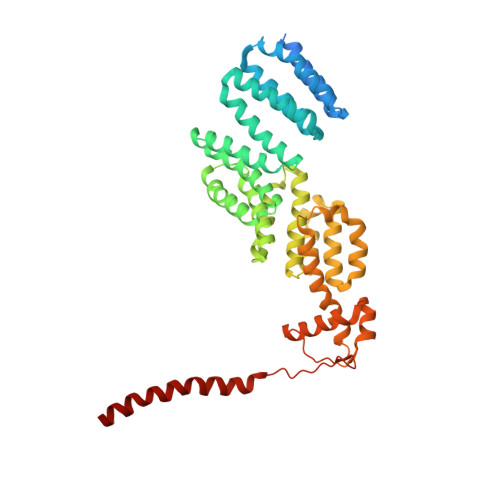
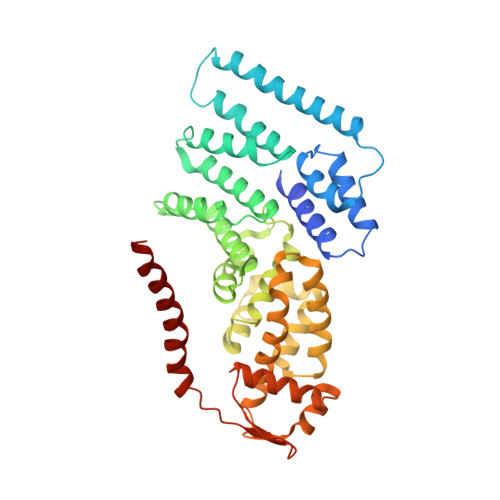
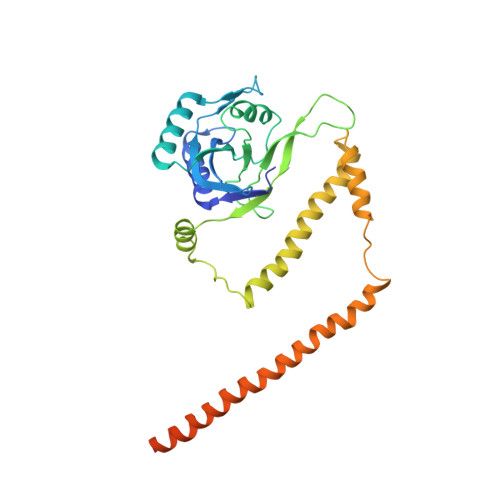
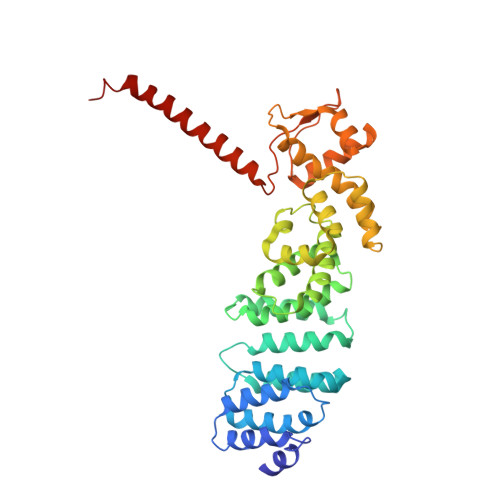
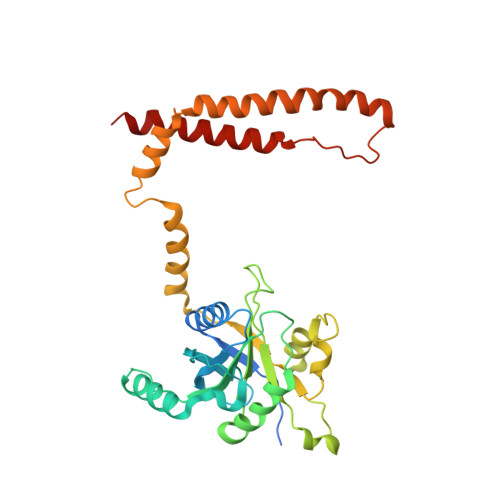
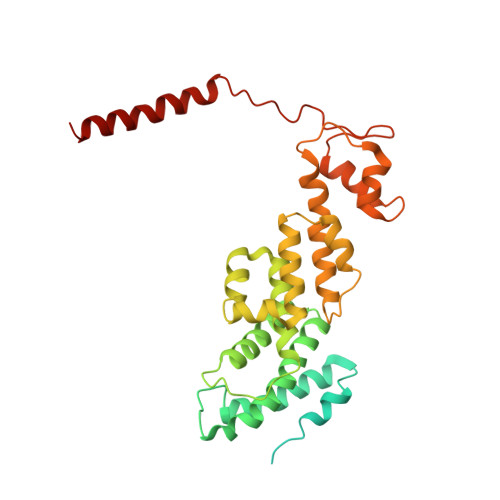
The proteasome is the major engine of protein degradation in all eukaryotic cells. At the heart of this machine is a heterohexameric ring of AAA (ATPases associated with diverse cellular activities) proteins that unfolds ubiquitylated target proteins that are concurrently translocated into a proteolytic chamber and degraded into peptides. Using cryoelectron microscopy, we determined a near-atomic-resolution structure of the 2.5-MDa human proteasome in its ground state, as well as subnanometer-resolution structures of the holoenzyme in three alternative conformational states. The substrate-unfolding AAA-ATPase channel is narrowed by 10 inward-facing pore loops arranged into two helices that run in parallel with each other, one hydrophobic in character and the other highly charged. The gate of the core particle was unexpectedly found closed in the ground state and open in only one of the alternative states. Coordinated, stepwise conformational changes of the regulatory particle couple ATP hydrolysis to substrate translocation and regulate gating of the core particle, leading to processive degradation.
- Center for Quantitative Biology, Peking University, Beijing 100871, China.
Organizational Affiliation: