New substrate analogue inhibitors of factor Xa containing 4-amidinobenzylamide as P1 residue: part 1.
Schweinitz, A., Sturzebecher, A., Sturzebecher, U., Schuster, O., Sturzebecher, J., Steinmetzer, T.(2006) Med Chem 2: 349-361
- PubMed: 16848746 Search on PubMed
- DOI: https://doi.org/10.2174/157340606777724040
- Primary Citation Related Structures:
5K0H - PubMed Abstract:
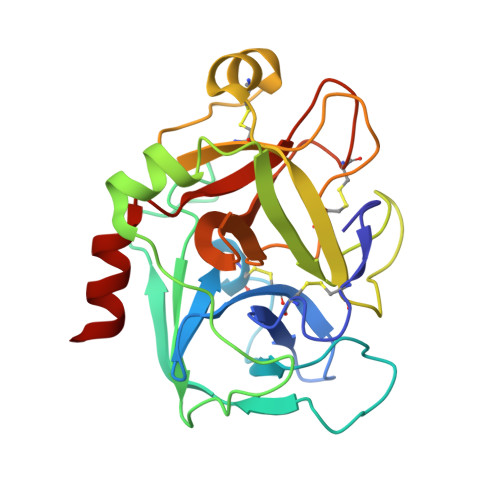
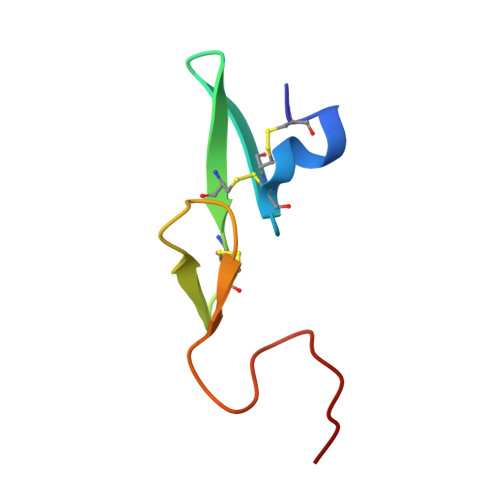
The trypsin-like serine protease factor Xa (fXa) is located at the convergence point of the intrinsic and extrinsic coagulation cascade, and therefore has emerged as an attractive target for the design of novel anticoagulants. During the development of substrate-analogue urokinase inhibitors we have found that the protection of the P3-dSer side chain leads to a scaffold of potent fXa inhibitors with the general structure R1-SO2-dSer(R2)-Gly-4-amidinobenzylamide. The first lead (3) with an N-terminal benzylsulfonyl group and dSer(tBu) as P3 residue inhibits human fXa with a Ki of 14 nM. A variety of derivatives with modified P4, P3, and P2 residues have been investigated in terms of inhibition of fXa and related proteases and for their anticoagulant potency and elimination behaviour. Most inhibitors were rapidly cleared from the circulation of rats. However, compound 48 (Ki= 3.5 nM), one of the most potent and selective inhibitors containing a dArg as P3 residue was relatively slowly eliminated (t1/2 approximately 1 h). Inhibitor 48 doubled clotting times in human plasma at 0.32 microM (aPTT) and 0.28 microM (PT), and is approximately 10-fold more potent than the reference fXa inhibitor DX-9065a in the inhibition of the prothrombinase complex. The structures of two inhibitors in complex with human fXa were solved by X-ray crystallography.
- Curacyte Chemistry GmbH, Winzerlaer Str. 2, D-07745 Jena, Germany.
Organizational Affiliation: