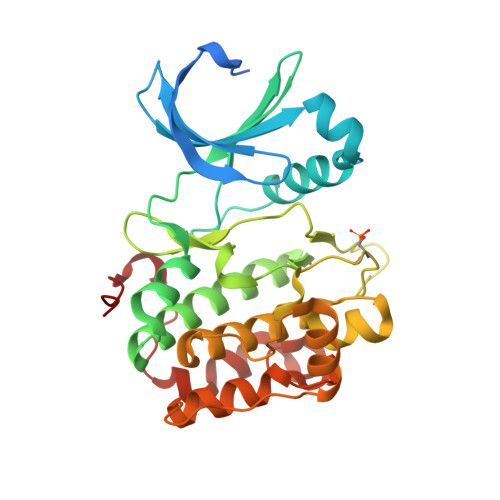
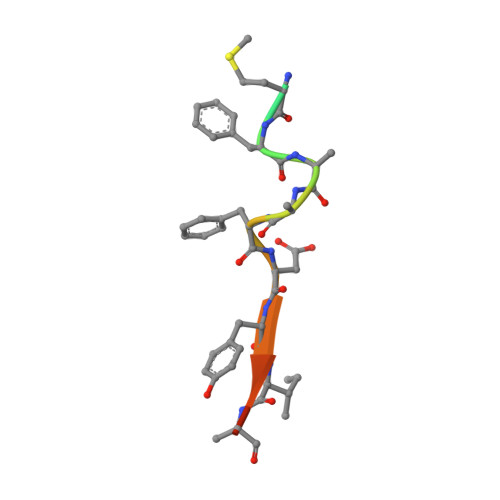
A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1.
Rettenmaier, T.J., Sadowsky, J.D., Thomsen, N.D., Chen, S.C., Doak, A.K., Arkin, M.R., Wells, J.A.(2014) Proc Natl Acad Sci U S A 111: 18590-18595
- PubMed: 25518860 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.1415365112
- Primary Citation Related Structures:
4RQK, 4RQV, 4RRV - PubMed Abstract:
There is great interest in developing selective protein kinase inhibitors by targeting allosteric sites, but these sites often involve protein-protein or protein-peptide interfaces that are very challenging to target with small molecules. Here we present a systematic approach to targeting a functionally conserved allosteric site on the protein kinase PDK1 called the PDK1-interacting fragment (PIF)tide-binding site, or PIF pocket. More than two dozen prosurvival and progrowth kinases dock a conserved peptide tail into this binding site, which recruits them to PDK1 to become activated. Using a site-directed chemical screen, we identified and chemically optimized ligand-efficient, selective, and cell-penetrant small molecules (molecular weight ∼ 380 Da) that compete with the peptide docking motif for binding to PDK1. We solved the first high-resolution structure of a peptide docking motif (PIFtide) bound to PDK1 and mapped binding energy hot spots using mutational analysis. We then solved structures of PDK1 bound to the allosteric small molecules, which revealed a binding mode that remarkably mimics three of five hot-spot residues in PIFtide. These allosteric small molecules are substrate-selective PDK1 inhibitors when used as single agents, but when combined with an ATP-competitive inhibitor, they completely suppress the activation of the downstream kinases. This work provides a promising new scaffold for the development of high-affinity PIF pocket ligands, which may be used to enhance the anticancer activity of existing PDK1 inhibitors. Moreover, our results provide further impetus for exploring the helix αC patches of other protein kinases as potential therapeutic targets even though they involve protein-protein interfaces.
- Chemistry and Chemical Biology Graduate Program, Department of Pharmaceutical Chemistry.
Organizational Affiliation: