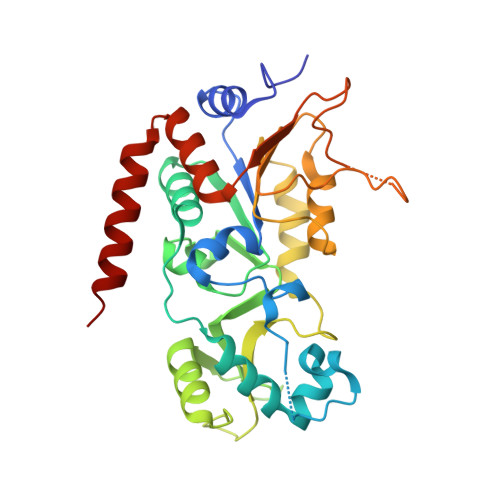
Selective Sirt2 inhibition by ligand-induced rearrangement of the active site.
Rumpf, T., Schiedel, M., Karaman, B., Roessler, C., North, B.J., Lehotzky, A., Olah, J., Ladwein, K.I., Schmidtkunz, K., Gajer, M., Pannek, M., Steegborn, C., Sinclair, D.A., Gerhardt, S., Ovadi, J., Schutkowski, M., Sippl, W., Einsle, O., Jung, M.(2015) Nat Commun 6: 6263-6263
- PubMed: 25672491 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/ncomms7263
- Primary Citation Related Structures:
4RMG, 4RMH, 4RMI, 4RMJ - PubMed Abstract:
Sirtuins are a highly conserved class of NAD(+)-dependent lysine deacylases. The human isotype Sirt2 has been implicated in the pathogenesis of cancer, inflammation and neurodegeneration, which makes the modulation of Sirt2 activity a promising strategy for pharmaceutical intervention. A rational basis for the development of optimized Sirt2 inhibitors is lacking so far. Here we present high-resolution structures of human Sirt2 in complex with highly selective drug-like inhibitors that show a unique inhibitory mechanism. Potency and the unprecedented Sirt2 selectivity are based on a ligand-induced structural rearrangement of the active site unveiling a yet-unexploited binding pocket. Application of the most potent Sirtuin-rearranging ligand, termed SirReal2, leads to tubulin hyperacetylation in HeLa cells and induces destabilization of the checkpoint protein BubR1, consistent with Sirt2 inhibition in vivo. Our structural insights into this unique mechanism of selective sirtuin inhibition provide the basis for further inhibitor development and selective tools for sirtuin biology.
- Institute of Pharmaceutical Sciences, Albert-Ludwigs-University Freiburg, Albertstraße 25, 79104 Freiburg im Breisgau, Germany.
Organizational Affiliation: