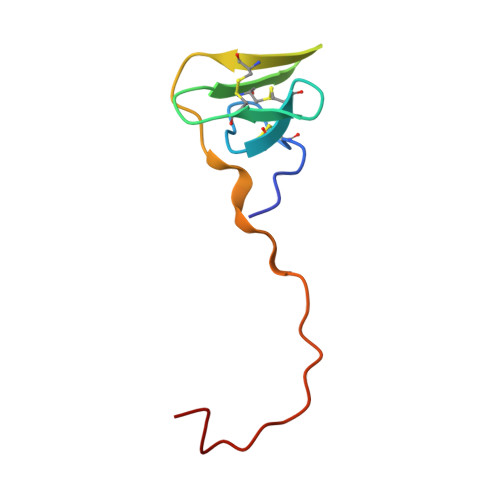
Refined structure of the hirudin-thrombin complex.
Rydel, T.J., Tulinsky, A., Bode, W., Huber, R.(1991) J Mol Biology 221: 583-601
- PubMed: 1920434 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(91)80074-5
- Primary Citation Related Structures:
4HTC - PubMed Abstract:
The structure of a recombinant hirudin (variant 2, Lys47) human alpha-thrombin complex has been refined using restrained least-squares methods to a crystallographic R-factor of 0.173. The hirudin structure consists of an N-terminal domain folded into a globular unit and a long 17-peptide C-terminal in an extended chain conformation. The N-terminal domain binds at the active-site of thrombin where Ile1' to Tyr3' penetrates to the catalytic triad. The alpha-amino group of Ile1' of hirudin makes a hydrogen bond with OG of Ser195 of thrombin, the side-chains of Ile1' and Tyr3' occupy the apolar site, Thr2' is at the entrance to, but does not enter, the S1 specificity site and Ile1' to Tyr3' form a parallel beta-strand with Ser214 to Gly219. The latter interaction is antiparallel in all other serine proteinase-protein inhibitor complexes. The extended C-terminal segment of hirudin, which is abundant in acidic residues, makes many electrostatic interactions with the fibrinogen binding exosite while the last five residues are in a 3(10) helical turn residing in a hydrophobic patch on the thrombin surface. The precision of the complementarity displayed by these two molecules produces numerous interactions, which although independently generally weak, together are responsible for the high degree of affinity and specificity. Although hirudin-thrombin and D-Phe-Pro-Arg-chloromethyl ketone-thrombin differ in conformation in the autolysis loop (Lys145 to Gly150), this is most likely due to different crystal packing interactions and changes in circular dichroism between the two are probably due to the inherent flexibility of the loop. An RGD sequence, which is generally known to be involved in cell surface receptor interactions, occurs in thrombin and is associated with a long solvent channel filled with water molecules leading to the surface from the end of the S1 site. However, the RGD triplet does not appear to be able to interact in concert in a surface binding mode.
- Department of Chemistry, Michigan State University, East Lansing 48824.
Organizational Affiliation: