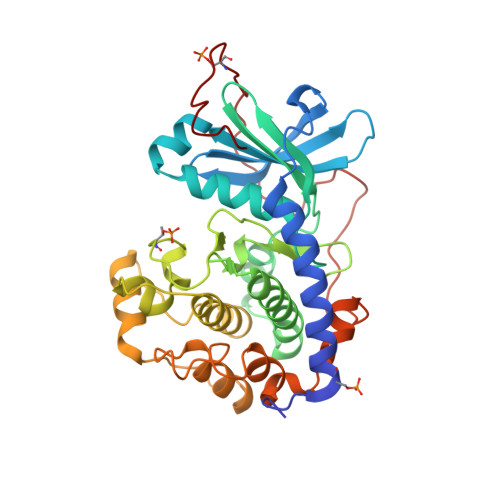
Anomalous dispersion analysis of inhibitor flexibility: a case study of the kinase inhibitor H-89
Pflug, A., Johnson, K.A., Engh, R.A.(2012) Acta Crystallogr Sect F Struct Biol Cryst Commun 68: 873-877
- PubMed: 22869112 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1107/S1744309112028655
- Primary Citation Related Structures:
3VQH - PubMed Abstract:
With its ability to show the interactions between drug-target proteins and small-molecule ligands, X-ray crystallography is an essential tool in drug-discovery programmes. However, its usefulness can be limited by crystallization artifacts or by the data resolution, and in particular when assumptions of unimodal binding (and isotropic motion) do not apply. Discrepancies between the modelled crystal structure and the physiological range of structures generally prevent quantitative estimation of binding energies. Improved crystal structure resolution will often not aid energy estimation because the conditions which provide the highest rigidity and resolution are not likely to reflect physiological conditions. Instead, strategies must be employed to measure and model flexibility and multiple binding modes to supplement crystallographic information. One useful tool is the use of anomalous dispersion for small molecules that contain suitable atoms. Here, an analysis of the binding of the kinase inhibitor H-89 to protein kinase A (PKA) is presented. H-89 contains a bromobenzene moiety that apparently binds with multiple conformations in the kinase ATP pocket. Using anomalous dispersion methods, it was possible to resolve these conformations into two distinct binding geometries.
- Norwegian Structural Biology Centre, Department of Chemistry, University of Tromsø, N-9037 Tromsø, Norway.
Organizational Affiliation: