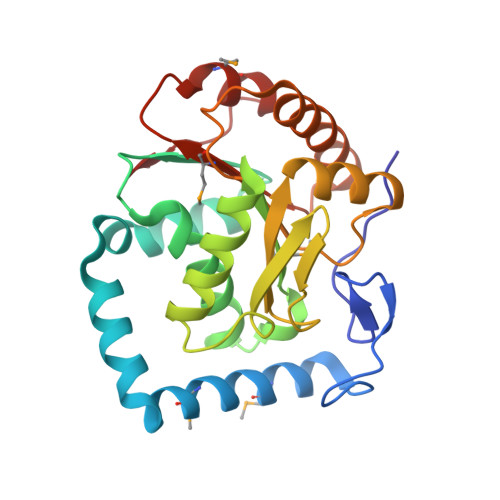
Crystal structure of human RNA helicase A (DHX9): structural basis for unselective nucleotide base binding in a DEAD-box variant protein.
Schutz, P., Wahlberg, E., Karlberg, T., Hammarstrom, M., Collins, R., Flores, A., Schuler, H.(2010) J Mol Biol 400: 768-782
- PubMed: 20510246 Search on PubMed
- DOI: https://doi.org/10.1016/j.jmb.2010.05.046
- Primary Citation Related Structures:
3LLM - PubMed Abstract:
RNA helicases of the DExD/H-box superfamily are critically involved in all RNA-related processes. No crystal structures of human DExH-box domains had been determined previously, and their structures were difficult to predict owing to the low level of homology among DExH-motif-containing proteins from diverse species. Here we present the crystal structures of the conserved domain 1 of the DEIH-motif-containing helicase DHX9 and of the DEAD-box helicase DDX20. Both contain a RecA-like core, but DHX9 differs from DEAD-box proteins in the arrangement of secondary structural elements and is more similar to viral helicases such as NS3. The N-terminus of the DHX9 core contains two long alpha-helices that reside on the surface of the core without contributing to nucleotide binding. The RNA-polymerase-II-interacting minimal transactivation domain sequence forms an extended loop structure that resides in a hydrophobic groove on the surface of the DEIH domain. DHX9 lacks base-selective contacts and forms an unspecific but important stacking interaction with the base of the bound nucleotide, and our biochemical analysis confirms that the protein can hydrolyze ATP, guanosine 5'-triphosphate, cytidine 5'-triphosphate, and uridine 5'-triphosphate. Together, these findings allow the localization of functional motifs within the three-dimensional structure of a human DEIH helicase and show how these enzymes can bind nucleotide with high affinity in the absence of a Q-motif.
- Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden.
Organizational Affiliation: