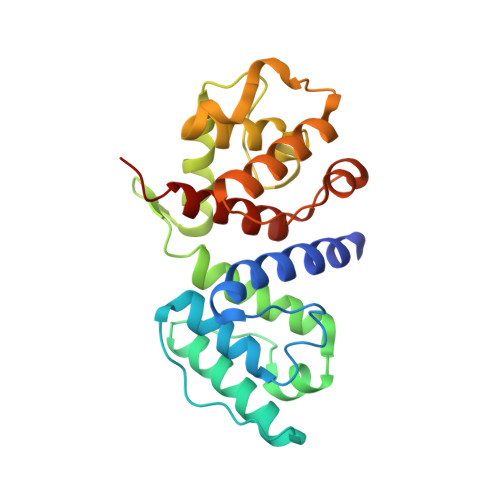
Crystal structure of the actin-binding domain of alpha-actinin-4 Lys255Glu mutant implicated in focal segmental glomerulosclerosis.
Lee, S.H., Weins, A., Hayes, D.B., Pollak, M.R., Dominguez, R.(2008) J Mol Biology 376: 317-324
- PubMed: 18164029 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1016/j.jmb.2007.11.084
- Primary Citation Related Structures:
2R0O - PubMed Abstract:
Mutations in alpha-actinin-4 have been linked to familial focal segmental glomerulosclerosis (FSGS), a common renal disorder in humans, and produce an apparent increase in the actin-binding affinity of alpha-actinin-4 in vitro. One of the mutations, in particular, Lys255Glu, falls in the middle of the actin-binding interface of the actin-binding domain (ABD). The ABD consists of tandem calponin homology (CH) domains (CH1 and CH2). The crystal structures of most ABDs display a compact conformation, characterized by extensive inter-CH interactions. However, the conformation of F-actin-bound ABDs is unsettled. Some electron microscopy studies find that the compact conformation is preserved upon binding to F-actin, whereas other studies suggest that the CHs separate and the ABD becomes extended. The Lys255Glu mutation in CH2 is significant in this regard since it removes a crucial inter-CH interaction with Trp147 of CH1, thought to stabilize the compact conformation. Together, the increased actin-binding affinity and the removal of this important inter-CH contact suggested that the Lys255Glu mutation might facilitate the transition toward the open ABD conformation proposed by some of the electron microscopy studies. However, the crystal structure of the ABD of alpha-actinin-4 Lys255Glu mutant described here displays the canonical compact conformation. Furthermore, the sedimentation coefficients by analytical ultracentrifugation of wild-type and FSGS mutant ABDs (Lys255Glu, Ser262Pro, and Thr259Ile) are nearly identical (2.50+/-0.03 S) and are in good agreement with the theoretical value calculated from the crystal structure (2.382 S), implying that the compact conformation is retained in solution. The absence of a structural change suggests that the compact ABD conformation observed in the majority of the structures is highly stable and is preserved in solution, even in FSGS mutant ABDs.
- University of Pennsylvania School of Medicine, Department of Physiology, 3700 Hamilton Walk, Philadelphia, PA 19104-6085, USA.
Organizational Affiliation: