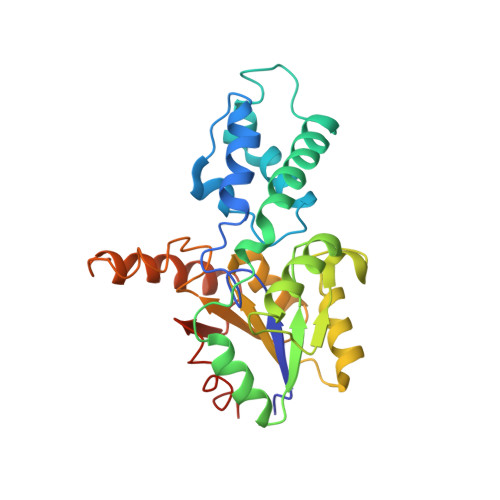
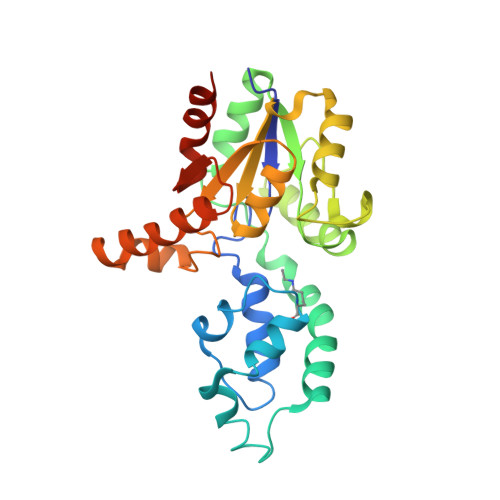
Diversification of function in the haloacid dehalogenase enzyme superfamily: The role of the cap domain in hydrolytic phosphoruscarbon bond cleavage.
Lahiri, S.D., Zhang, G., Dunaway-Mariano, D., Allen, K.N.(2006) Bioorg Chem 34: 394-409
- PubMed: 17070898 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1016/j.bioorg.2006.09.007
- Primary Citation Related Structures:
2IOF, 2IOH - PubMed Abstract:
Phosphonatase functions in the 2-aminoethylphosphonate (AEP) degradation pathway of bacteria, catalyzing the hydrolysis of the C-P bond in phosphonoacetaldehyde (Pald) via formation of a bi-covalent Lys53ethylenamine/Asp12 aspartylphosphate intermediate. Because phosphonatase is a member of the haloacid dehalogenase superfamily, a family predominantly comprised of phosphatases, the question arises as to how this new catalytic activity evolved. The source of general acid-base catalysis for Schiff-base formation and aspartylphosphate hydrolysis was probed using pH-rate profile analysis of active-site mutants and X-ray crystallographic analysis of modified forms of the enzyme. The 2.9 A X-ray crystal structure of the mutant Lys53Arg complexed with Mg2+ and phosphate shows that the equilibrium between the open and the closed conformation is disrupted, favoring the open conformation. Thus, proton dissociation from the cap domain Lys53 is required for cap domain-core domain closure. The likely recipient of the Lys53 proton is a water-His56 pair that serves to relay the proton to the carbonyl oxygen of the phosphonoacetaldehyde (Pald) substrate upon addition of the Lys53. The pH-rate profile analysis of active-site mutants was carried out to test this proposal. The proximal core domain residues Cys22 and Tyr128 were ruled out, and the role of cap domain His56 was supported by the results. The X-ray crystallographic structure of wild-type phosphonatase reduced with NaBH4 in the presence of Pald was determined at 2.4A resolution to reveal N epsilon-ethyl-Lys53 juxtaposed with a sulfate ligand bound in the phosphate site. The position of the C2 of the N-ethyl group in this structure is consistent with the hypothesis that the cap domain N epsilon-ethylenamine-Lys53 functions as a general base in the hydrolysis of the aspartylphosphate bi-covalent enzyme intermediate. Because the enzyme residues proposed to play a key role in P-C bond cleavage are localized on the cap domain, this domain appears to have evolved to support the diversification of the HAD phosphatase core domain for catalysis of hydrolytic P-C bond cleavage.
- Department of Physiology and Biophysics, Boston University School of Medicine, Boston, MA 02118-2394, USA.
Organizational Affiliation: