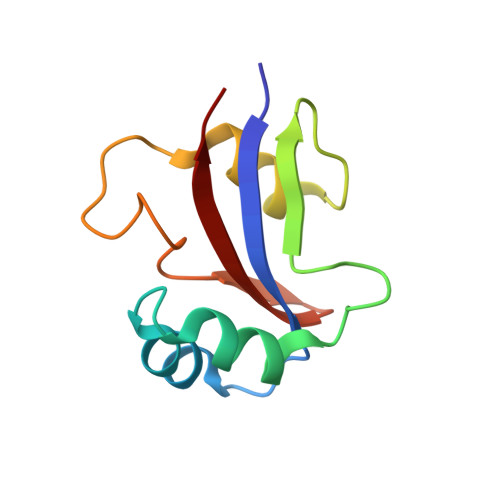
NMR solution structure of hPar14 reveals similarity to the peptidyl prolyl cis/trans isomerase domain of the mitotic regulator hPin1 but indicates a different functionality of the protein.
Sekerina, E., Rahfeld, J.U., Muller, J., Fanghanel, J., Rascher, C., Fischer, G., Bayer, P.(2000) J Mol Biology 301: 1003-1017
- PubMed: 10966801 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.2000.4013
- Primary Citation Related Structures:
1EQ3 - PubMed Abstract:
The 131-amino acid residue parvulin-like human peptidyl-prolyl cis/trans isomerase (PPIase) hPar14 was shown to exhibit sequence similarity to the regulator enzyme for cell cycle transitions human hPin1, but specificity for catalyzing pSer(Thr)-Pro cis/trans isomerizations was lacking. To determine the solution structure of hPar14 the (1)H, (13)C, and (15)N chemical shifts of this protein have been assigned using heteronuclear two and three-dimensional NMR experiments on unlabeled and uniformly (15)N/(13)C-labeled recombinant protein isolated from Escherichia coli cells that overexpress the protein. The chemical shift assignments were used to interpret the NOE data, which resulted in a total of 1042 NOE restraints. The NOE restraints were used along with 71 dihedral angle restraints and 38 hydrogen bonding restraints to produce 50 low-energy structures. The hPar14 folds into a betaalpha(3)betaalphabeta(2) structure, and contains an unstructured 35-amino acid basic tail N-terminal to the catalytic core that replaces the WW domain of hPin1 homologs. The three-dimensional structures of hPar14 and the PPIase domain of human hPin1 reveal a high degree of conservation. The root-mean-square deviations of the mean atomic coordinates of the heavy atoms of the backbone between residues 38 to 45, 50 to 58, 64 to 70, 81 to 86, 115 to 119 and 122 to 128 of hPar14 were 0.81(+/-0.07) A. The hPar14 model structure provides insight into how this class of PPIases may select preferential secondary catalytic sites, and also allows identification of a putative DNA-binding motif in parvulin-like PPIases.
- Max-Planck-Stelle for Enzymology of Protein Folding, 06120 Halle/Saale, Weinberg 22, Germany.
Organizational Affiliation: