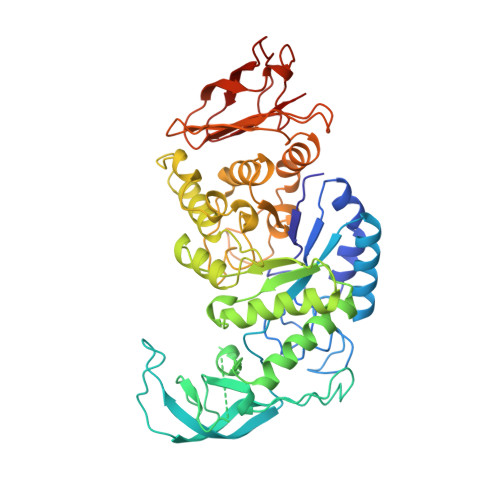
Crystal structure of thermostable alpha-amylase from Bacillus licheniformis refined at 1.7 A resolution
Hwang, K.Y., Song, H.K., Chang, C., Lee, J., Lee, S.Y., Kim, K.K., Choe, S., Sweet, R.M., Suh, S.W.(1997) Mol Cells 7: 251-258
- PubMed: 9163741 Search on PubMed
- Primary Citation Related Structures:
1VJS - PubMed Abstract:
alpha-Amylases (alpha-1,4-glucan-4-glucanohydrolase, E.C.3.2.1.1) catalyze the cleavage of alpha-1, 4-glucosidic linkages of starch components, glycogen, and various oligosaccharides. Thermostable alpha-amylases from Bacillus species are of great industrial importance in the production of corn syrup or dextrose. Thermostable alpha-amylase from Bacillus licheniformis, a monomeric enzyme with molecular mass of 55,200 Da (483 amino acid residues), shows a remarkable heat stability. This enzyme provides an attractive model for investigating the structural basis for thermostability of proteins. The three-dimensional structure of thermostable alpha-amylase from Bacillus licheniformis has been determined by the multiple isomorphous replacement method of X-ray crystallography. The structure has been refined to a crystallographic R-factor of 19.9% for 58,601 independent reflections with F0 > 2 sigma F0 between 8.0 and 1.7 A resolution, with root mean square deviations of 0.013 A from ideal bond lengths and 1.72 degrees from ideal bond angles. The final model consists of 469 amino acid residues and 294 water molecules. Missing from the model are the N- and C-termini and the segment between Trp182 and Asn192. Like other alpha-amylases, the polypeptide chain folds into three distinct domains. The first domain (domain A), consisting of 291 residues (from residue 3 to 103 and 207 to 396), forms a (beta/alpha)8-barrel structure. The second domain (domain B), consisting of residues 104 to 206, is inserted between the third beta-strand and the third alpha-helix of domain A. The third C-terminal domain (domain C), consisting of residues 397 to 482, folds into an eight-stranded antiparallel beta-barrel. Neither calcium ion nor chloride ion is located near the active site. This study reveals the architecture of the thermostable alpha-amylase from Bacillus licheniformis. By homology with other alpha-amylases, important active site residues can be identified as Asp231, Glu261, and Asp328, which are all located at the C-terminal end of the central (beta/alpha)8-barrel. Since many of the stabilizing and destabilizing mutations obtained so far fall in domain B or at its border, this region of the enzyme appears to be important for thermostability. The factors responsible for the remarkable thermostability of this enzyme may be increased ionic interactions, reduced surface area, and increased packing interactions in the interior.
- Department of Chemistry, Seoul National University, Korea.
Organizational Affiliation: