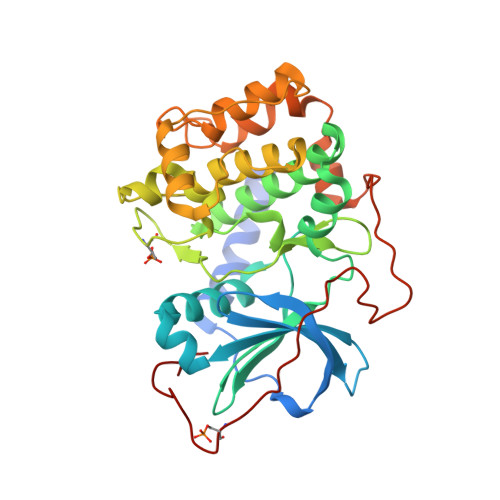
Mutants of protein kinase A that mimic the ATP-binding site of protein kinase B (AKT)
Gassel, M., Breitenlechner, C.B., Rueger, P., Jucknischke, U., Schneider, T., Huber, R., Bossemeyer, D., Engh, R.A.(2003) J Mol Biol 329: 1021-1034
- PubMed: 12798691 Search on PubMed
- DOI: https://doi.org/10.1016/s0022-2836(03)00518-7
- Primary Citation Related Structures:
1Q24, 1Q61, 1Q62 - PubMed Abstract:
The mutation of well behaved enzymes in order to simulate less manageable cognates is the obvious approach to study specific features of the recalcitrant target. Accordingly, the prototypical protein kinase PKA serves as a model for many kinases, including the closely related PKB, an AGC family protein kinase now implicated as oncogenic in several cancers. Two residues that differ between the alpha isoforms of PKA and PKB at the adenine-binding site generate differing shapes of the binding surface and are likely to play a role in ligand selectivity. As the corresponding mutations in PKA, V123A would enlarge the adenine pocket, while L173M would alter both the shape and its electronic character of the adenine-binding surface. We have determined the structures of the corresponding double mutant (PKAB2: PKAalpha V123A, L173M) in apo and MgATP-bound states, and observed structural alterations of a residue not previously involved in ATP-binding interactions: the side-chain of Q181, which in native PKA points away from the ATP-binding site, adopts in apo double mutant protein a new rotamer conformation, which places the polar groups at the hinge region in the ATP pocket. MgATP binding forces Q181 back to the position seen in native PKA. The crystal structure shows that ATP binding geometry is identical with that in native PKA but in this case was determined under conditions with only a single Mg ion ligand. Surface plasmon resonance spectroscopy studies show that significant energy is required for this ligand-induced transition. An additional PKA/PKB mutation, Q181K, corrects the defect, as shown both by the crystal structure of triple mutant PKAB3 (PKAalpha V123A, L173M, Q181K) and by surface plasmon resonance spectroscopy binding studies with ATP and three isoquinoline inhibitors. Thus, the triple mutant serves well as an easily crystallizable model for PKB inhibitor interactions. Further, the phenomenon of Q181 shows how crystallographic analysis should accompany mutant studies to monitor possible spurious structural effects.
- German Cancer Research Center (DKFZ), Division of Pathochemistry, Im Neuenheimer Feld 280, 69120 Heidelberg, Germany.
Organizational Affiliation: