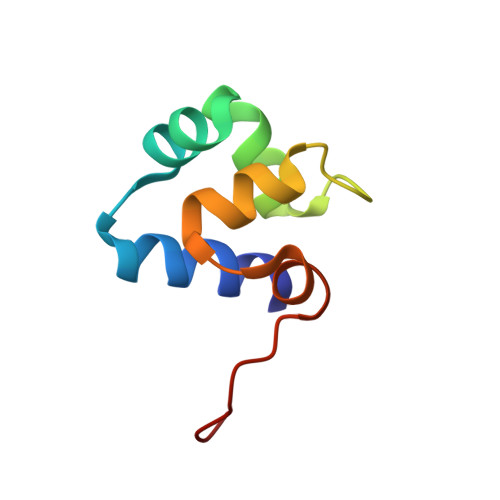
Determination of the nuclear magnetic resonance solution structure of the DNA-binding domain (residues 1 to 69) of the 434 repressor and comparison with the X-ray crystal structure.
Neri, D., Billeter, M., Wuthrich, K.(1992) J Mol Biol 223: 743-767
- PubMed: 1311771 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(92)90987-u
- Primary Citation Related Structures:
1PRA - PubMed Abstract:
The DNA-binding domain of the phage 434 repressor consisting of N-terminal residues 1 to 69 (434 repressor(1-69)), was expressed in Escherichia coli with natural isotope abundance, uniform 15N-labeling and biosynthetically directed fractional 13C-labeling in extent of about 10%. With these protein preparations the three-dimensional structure was determined in solution. The techniques used were nuclear magnetic resonance (n.m.r.) spectroscopy for the collection of conformational constraints, calculation of the protein structure from the n.m.r. data with the program DIANA and structure refinements by restrained energy minimization with a modified version of the program AMBER. A group of 20 conformers characterizes a well-defined structure for residues 1 to 63, with an average of 0-6 A for the root-mean-square deviations (RMSD) calculated for the backbone atoms of the individual conformers relative to the mean co-ordinates. The spatial structure of C-terminal residues 64 to 69 is not defined by the n.m.r. data. The molecular architecture of the 434 repressor(1-69) in solution includes five alpha-helices extending from residues 2 to 13, 17 to 24, 28 to 35, 45 to 52 and 56 to 60, which enclose a well-defined hydrophobic core. The n.m.r. structure is closely similar to the reported crystal structure of the 434 repressor(1-69), with an RMSD value of 1.1 A for the backbone atoms of residues 1 to 63. Small differences between the two structures in regions of the first helix and the loop between helices 3 and 4 were analyzed relative to possible correlations with protein-protein contacts in the crystal lattice and the different milieus of pH and ionic strength in the crystals and n.m.r. samples. Further systematic comparisons of local conformational features indicated that there are correlations between amino acid types, local precision of the structure determination by both techniques and local differences between the structures in the crystals and in solution. Overall, hydrophobic residues are most precisely characterized and agree most closely in the two environments.
- Institut für Molekularbiologie und Biophysik Eidgenössische Technische Hochschule-Hönggerberg, Zürich, Switzerland.
Organizational Affiliation: