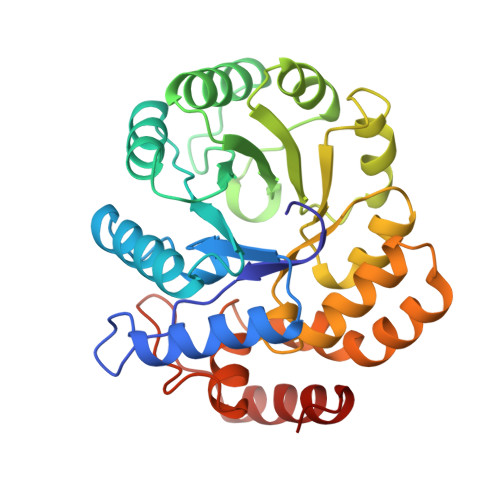
The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 A resolution.
Mirwaldt, C., Korndorfer, I., Huber, R.(1995) J Mol Biology 246: 227-239
- PubMed: 7853400 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.1994.0078
- Primary Citation Related Structures:
1DHP - PubMed Abstract:
The crystal structure of dihydrodipicolinate synthase from E. coli was determined by multiple isomorphous replacement methods. The structure was refined at a resolution of 2.5 A and the final R-factor is 19.6% for 32,190 reflections between 10.0 A and 2.5 A and F > 2 sigma (F). The crystallographic asymmetric unit contains two monomers related by approximate 2-fold symmetry. A tetramer with approximate 222 symmetry is built up by crystallographic symmetry. The tetramer is almost planar with no contacts between the subunits related by the non-crystallographic dyad. The active sites are accessible from a wide water-filled channel in the center of the tetramer. The dihydrodipicolinate synthase monomer is composed of two domains. Each polypeptide chain is folded into an 8-fold alpha/beta barrel and a C-terminal alpha-helical domain comprising residues 224 to 292. The fold is similar to that of N-acetylneuraminate lyase. The active site lysine 161 is located in the alpha/beta barrel and has access via two entrances from the C-terminal side of the barrel.
- Max-Planck Institut für Biochemie, Martinsried, FRG.
Organizational Affiliation: