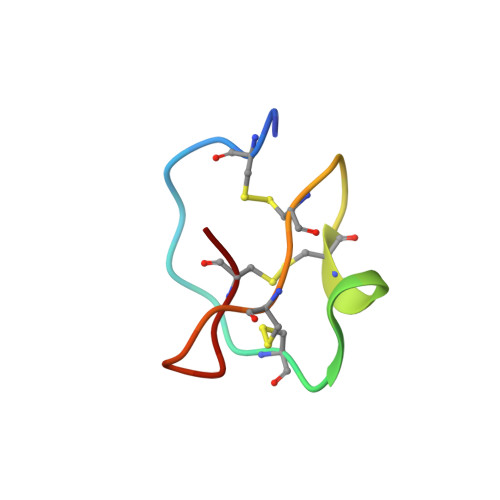
Determination of the complete three-dimensional structure of the trypsin inhibitor from squash seeds in aqueous solution by nuclear magnetic resonance and a combination of distance geometry and dynamical simulated annealing.
Holak, T.A., Gondol, D., Otlewski, J., Wilusz, T.(1989) J Mol Biol 210: 635-648
- PubMed: 2614837 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(89)90137-x
- Primary Citation Related Structures:
1CTI, 2CTI - PubMed Abstract:
The complete three-dimensional structure of the trypsin inhibitor from seeds of the squash Cucurbita maxima in aqueous solution was determined on the basis of 324 interproton distance constraints, 80 non-nuclear Overhauser effect distances, and 22 hydrogen-bonding constraints, supplemented by 27 phi backbone angle constraints derived from nuclear magnetic resonance measurements. The nuclear magnetic resonance input data were converted to the distance constraints in a semiquantitative manner after a sequence specific assignment of 1H spectra was obtained using two-dimensional nuclear magnetic resonance techniques. Stereospecific assignments were obtained for 17 of the 48 prochiral centers of the squash trypsin inhibitor using the floating chirality assignment introduced at the dynamical simulated annealing stage of the calculations. A total of 34 structures calculated by a hybrid distance geometry-dynamical simulated annealing method exhibit well-defined positions for both backbone and side-chain atoms. The average atomic root-mean-square difference between the individual structures and the minimized mean structure is 0.35(+/- 0.08) A for the backbone atoms and 0.89(+/- 0.17) A for all heavy atoms. The precision of the structure determination is discussed and correlated to the experimental input data.
- Max-Planck-Institut für Biochemie, Martinsried bei München, F.R.G.
Organizational Affiliation: