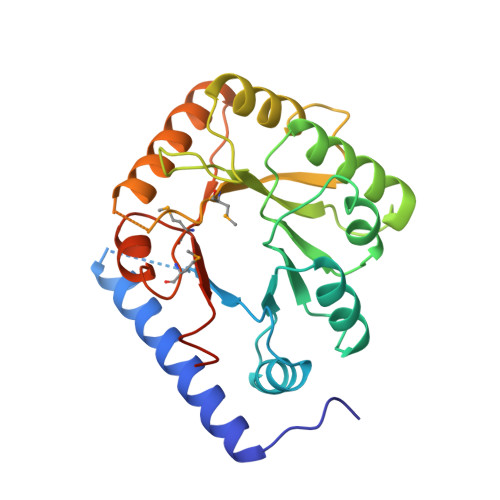
Structure of a yeast hypothetical protein selected by a structural genomics approach.
Eswaramoorthy, S., Gerchman, S., Graziano, V., Kycia, H., Studier, F.W., Swaminathan, S.(2003) Acta Crystallogr D Biol Crystallogr 59: 127-135
- PubMed: 12499548 Search on PubMed
- DOI: https://doi.org/10.1107/s0907444902018012
- Primary Citation Related Structures:
1B54, 1CT5 - PubMed Abstract:
Yeast hypothetical protein YBL036C (SWISS-PROT P38197), initially thought to be a member of an 11-protein family, was selected for crystal structure determination since no structural or functional information was available. The structure has been determined independently by MIR and MAD methods to 2.0 A resolution. The MAD structure was determined largely through automated model building. The protein folds as a TIM barrel beginning with a long N-terminal helix, in contrast to the classic triose phosphate isomerase (TIM) structure, which begins with a beta-strand. A cofactor, pyridoxal 5'-phosphate, is covalently bound near the C-terminal end of the barrel, the usual active site in TIM-barrel folds. A single-domain monomeric molecule, this yeast protein resembles the N-terminal domain of alanine racemase or ornithine decarboxylase, both of which are two-domain dimeric proteins. The yeast protein has been shown to have amino-acid racemase activity. Although selected as a member of a protein family having no obvious relationship to proteins of known structure, the protein fold turned out to be a well known and widely distributed fold. This points to the need for a more comprehensive base of structural information and better structure-modeling tools before the goal of structure prediction from amino-acid sequences can be realised. In this case, similarity to a known structure allowed inferences to be made about the structure and function of a widely distributed protein family.
- Biology Department, Brookhaven National Laboratory, Upton, NY 11973, USA.
Organizational Affiliation: