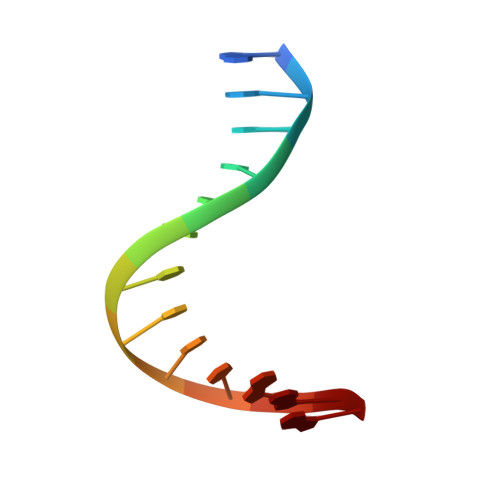
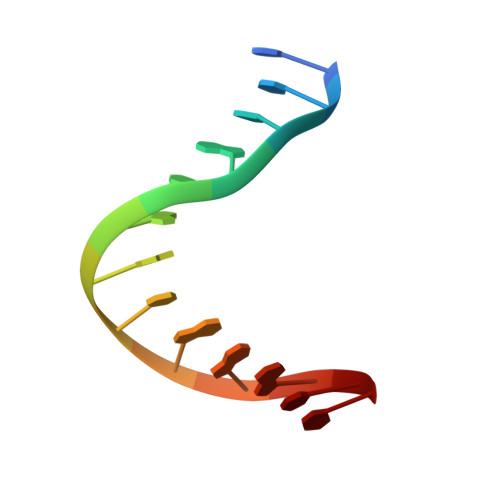
Solution structure of a conserved DNA sequence from the HIV-1 genome: restrained molecular dynamics simulation with distance and torsion angle restraints derived from two-dimensional NMR spectra.
Mujeeb, A., Kerwin, S.M., Kenyon, G.L., James, T.L.(1993) Biochemistry 32: 13419-13431
- PubMed: 8257678 Search on PubMed
- DOI: https://doi.org/10.1021/bi00212a007
- Primary Citation Related Structures:
140D, 141D, 142D - PubMed Abstract:
The three-dimensional solution structure of a trisdecamer DNA duplex sequence, d(AGCTTGCCTTGAG).d(CTCAAGGCAAGCT), from a conserved region of HIV-1 genome's long terminal repeat, has been investigated using NMR spectroscopy and restrained molecular dynamics calculations. Interproton distances derived from two-dimensional nuclear Overhauser enhancement (2D NOE) experiments, using the iterative complete relaxation matrix algorithm MARDIGRAS, and torsion angles for sugar rings, estimated from stimulated fitting of double-quantum-filtered correlation (2QF-COSY) spectra, were obtained [Mujeeb, A., Kerwin, S. M., Egan, W. M., Kenyon, G. L., & James, T. L. (1992) Biochemistry 31, 9325-38]. These structural restraints have now been employed as the basis for structure refinement using restrained molecular dynamics (rMD) to search conformational space for structures consistent with the experimental restraints. Specifically, upper and lower bounds on the restraints were incorporated into the AMBER (version 4.0) total potential energy function of the system, the bounds being used to define the width of a flat-well penalty term in the AMBER force field. Confidence in the time-averaged structure obtained is engendered by convergence to essentially the same structure (root-mean-square deviation approximately 0.9 A) when two quite different DNA models, A-DNA and B-DNA (RMSD approximately 6.5 A), were employed as starting structures and when various initial trajectories were used for the rMD runs. The derived structure is further supported by the total energy calculated, the restraint violation energy, the restraint deviations, and the fit with experimental data. For the latter, the sixth-root residual index indicated a good fit of the determined structure with experimental 2D NOE spectral intensities (R1x < 0.07), and the RMS difference between vicinal proton coupling constants calculated for the derived structure and experimental coupling constants were also in reasonable agreement (JRMS = 0.9 Hz). While the structure of the trisdecamer is basically in the B-DNA family, some structural parameters manifest interesting local variations. The helix parameters indicate that, compared with classical B-DNA, the structure is longitudinally more compressed. Local structural variations at the two TG steps in particular together create bending into the major groove of the duplex. Comparison of the two-CTTG-tetrads in the duplex reveals that they have similar structures, with the TT moieties being almost identical; however, the -CTTG-pur sequence has a larger roll and slide for the -TG- step than for the -CTTG-pyr sequence, in accord with published X-ray crystallographic conclusions.
- Department of Pharmaceutical Chemistry, University of California, San Francisco 94143-0446.
Organizational Affiliation: