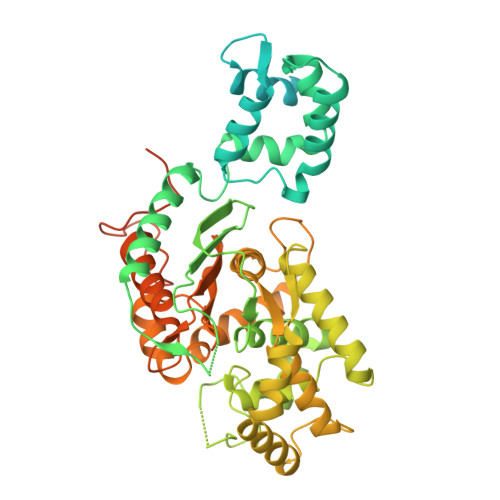
Structure and enzymology of glutaminase S482C and H461L variants associated with excess brain glutamate and neurological disease.
Crane, C.S., McIssac, T.K., Milano, S.K., Cerione, R.A., Ulrich, S.M.(2026) J Biological Chem 302: 113091-113091
- PubMed: 42055345 Search on PubMed
- DOI: https://doi.org/10.1016/j.jbc.2026.113091
- Primary Citation Related Structures:
9PIA - PubMed Abstract:
Glutaminase (GLS) catalyzes the hydrolysis of glutamine to produce glutamate, the brain's principal excitatory neurotransmitter. Two de novo gain-of-function mutations in GLS, S482C and H461L, were recently identified in patients with developmental delay, epilepsy, and infantile cataract. These patients exhibited high glutamate and low glutamine concentrations in the brain, suggesting that the GLS variants have abnormal enzymology. Here, we examined the enzymatic properties of the mutant enzymes and found that they no longer require the anionic activator phosphate to stimulate enzymatic activity or induce filament formation. The mutant enzymes also exhibit a total (S482C) or partial (H461L) loss of glutamate product inhibition, lifting this restriction on glutamate accumulation. Structural analysis of the S482C variant shows the mutation shifts the key catalytic residue Y466 into its catalytically active configuration and disrupts a key hydrogen bond between Y466 and the glutamate product, explaining how the S482C variant has enzymatic activity in the absence of phosphate and is insensitive to glutamate product inhibition. These results shed new light on the mechanism of phosphate activation and glutamate product inhibition of GLS and show that loss of these enzymatic properties disrupts glutamate homeostasis in the brain and causes neurological disease.
- Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853.
Organizational Affiliation: