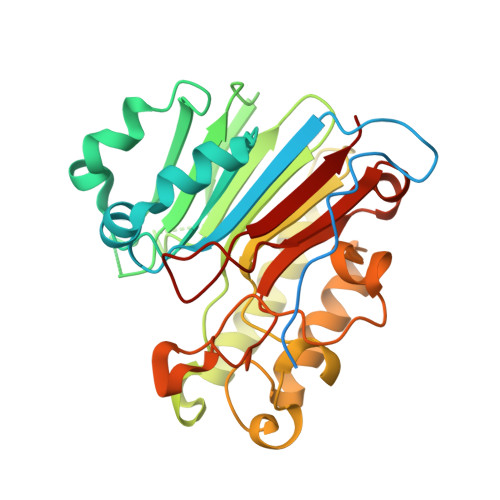
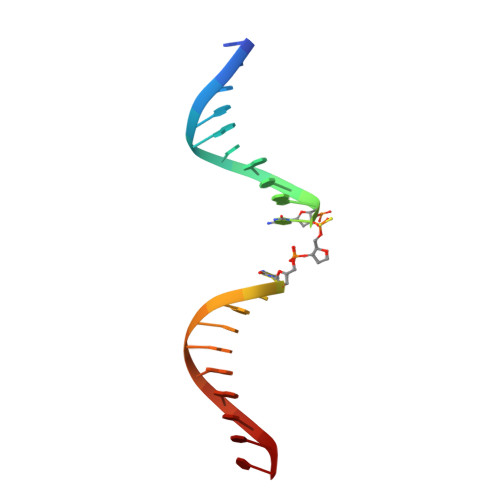
Apurinic/apyrimidinic (AP) endonuclease 1 processing of AP sites with 5' mismatches.
Fairlamb, M.S., Whitaker, A.M., Freudenthal, B.D.(2018) Acta Crystallogr D Struct Biol 74: 760-768
- PubMed: 30082511 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1107/S2059798318003340
- Primary Citation Related Structures:
6BOQ, 6BOR, 6BOS, 6BOT, 6BOU, 6BOV, 6BOW - PubMed Abstract:
Despite the DNA duplex being central to biological functions, many intricacies of this molecule, including the dynamic nature of mismatched base pairing, are still unknown. The unique conformations adopted by DNA mismatches can provide insight into the forces at play between nucleotides. Moreover, DNA-binding proteins apply their own individualized steric and electrochemical influences on the nucleotides that they interact with, further altering base-pairing conformations. Here, seven X-ray crystallographic structures of the human nuclease apurinic/apyrimidinic (AP) endonuclease 1 (APE1) in complex with its substrate target flanked by a 5' mismatch are reported. The structures reveal how APE1 influences the conformations of a variety of different mismatched base pairs. Purine-purine mismatches containing a guanine are stabilized by a rotation of the guanine residue about the N-glycosidic bond to utilize the Hoogsteen edge for hydrogen bonding. Interestingly, no rotation of adenine, the other purine, is observed. Mismatches involving both purine and pyrimidine bases adopt wobble conformations to accommodate the mismatch. Pyrimidine-pyrimidine mismatches also wobble; however, the smaller profile of a pyrimidine base results in a gap between the Watson-Crick faces that is reduced by a C1'-C1' compression. These results advance our understanding of mismatched base pairing and the influence of a bound protein.
- Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, KS 66016, USA.
Organizational Affiliation: