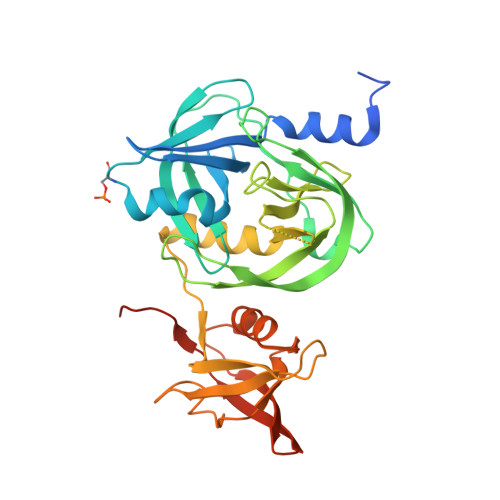
The crystal structure of an essential high-temperature requirement protein HtrA1 (Rv1223) from Mycobacterium tuberculosis reveals its unique features.
Singh, K.H., Yadav, S., Kumar, D., Biswal, B.K.(2018) Acta Crystallogr D Struct Biol 74: 906-921
- PubMed: 30198900 Search on PubMed
- DOI: https://doi.org/10.1107/S205979831800952X
- Primary Citation Related Structures:
5ZVJ - PubMed Abstract:
High-temperature requirement A (HtrA) proteins, which are members of the heat-shock-induced serine protease family, are involved in extracytoplasmic protein quality control and bacterial survival strategies under stress conditions, and are associated with the virulence of several pathogens; they are therefore major drug targets. Mycobacterium tuberculosis possesses three putative HtrAs: HtrA1 (Rv1223), HtrA2 (Rv0983) and HtrA3 (Rv0125). Each has a cytoplasmic region, a transmembrane helix and a periplasmic region. Here, the crystal structure of the periplasmic region consisting of a protease domain (PD) and a PDZ domain from an M. tuberculosis HtrA1 mutant (mHtrA1 S387A ) is reported at 2.7 Å resolution. Although the mHtrA1 S387A PD shows structural features similar to those of other HtrAs, its loops, particularly L3 and LA, display different conformations. Loop L3 communicates between the PDs of the trimer and the PDZ domains and undergoes a transition from an active to an inactive conformation, as reported for an equivalent HtrA (DegS). Loop LA, which is responsible for higher oligomer formation owing to its length (50 amino acids) in DegP, is very short in mHtrA1 S387A (five amino acids), as in mHtrA2 (also five amino acids), and therefore lacks essential interactions for the formation of higher oligomers. Notably, a well ordered loop known as the insertion clamp in the PDZ domain interacts with the protease domain of the adjacent molecule, which possibly aids in the stabilization of a trimeric functional unit of this enzyme. The three-dimensional structure of mHtrA1 S387A presented here will be useful in the design of enzyme-specific antituberculosis inhibitors.
- Structural and Functional Biology Laboratory, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110 067, India.
Organizational Affiliation: