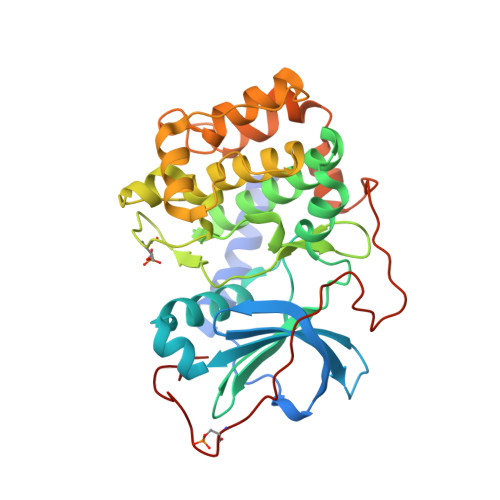
Accounting for Conformational Variability in Protein-Ligand Docking with NMR-Guided Rescoring
Skjaerven, L., Codutti, L., Angelini, A., Grimaldi, M., Latek, D., Monecke, P., Dreyer, M.K., Carlomagno, T.(2013) J Am Chem Soc 135: 5819-5827
- PubMed: 23565800 Search on PubMed
- DOI: https://doi.org/10.1021/ja4007468
- Primary Citation Related Structures:
4IE9, 4IJ9 - PubMed Abstract:
A key component to success in structure-based drug design is reliable information on protein-ligand interactions. Recent development in NMR techniques has accelerated this process by overcoming some of the limitations of X-ray crystallography and computational protein-ligand docking. In this work we present a new scoring protocol based on NMR-derived interligand INPHARMA NOEs to guide the selection of computationally generated docking modes. We demonstrate the performance in a range of scenarios, encompassing traditionally difficult cases such as docking to homology models and ligand dependent domain rearrangements. Ambiguities associated with sparse experimental information are lifted by searching a consensus solution based on simultaneously fitting multiple ligand pairs. This study provides a previously unexplored integration between molecular modeling and experimental data, in which interligand NOEs represent the key element in the rescoring algorithm. The presented protocol should be widely applicable for protein-ligand docking also in a different context from drug design and highlights the important role of NMR-based approaches to describe intermolecular ligand-receptor interactions.
- EMBL, Structural and Computational Biology Unit, Meyerhofstrasse 1, D-69117 Heidelberg, Germany.
Organizational Affiliation: