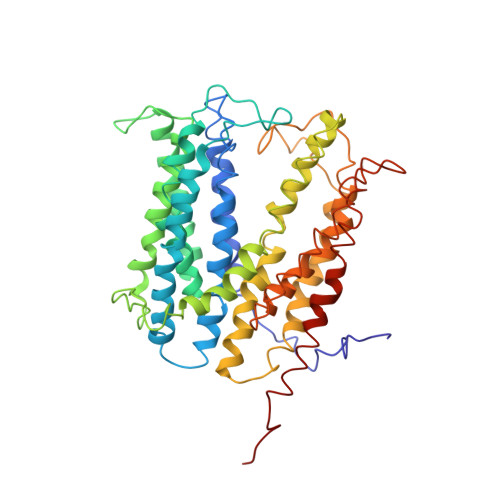
Structures of a Na+-coupled, substrate-bound MATE multidrug transporter.
Lu, M., Symersky, J., Radchenko, M., Koide, A., Guo, Y., Nie, R., Koide, S.(2013) Proc Natl Acad Sci U S A 110: 2099-2104
- PubMed: 23341609 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.1219901110
- Primary Citation Related Structures:
4HUK, 4HUL, 4HUM, 4HUN - PubMed Abstract:
Multidrug transporters belonging to the multidrug and toxic compound extrusion (MATE) family expel dissimilar lipophilic and cationic drugs across cell membranes by dissipating a preexisting Na(+) or H(+) gradient. Despite its clinical relevance, the transport mechanism of MATE proteins remains poorly understood, largely owing to a lack of structural information on the substrate-bound transporter. Here we report crystal structures of a Na(+)-coupled MATE transporter NorM from Neisseria gonorrheae in complexes with three distinct translocation substrates (ethidium, rhodamine 6G, and tetraphenylphosphonium), as well as Cs(+) (a Na(+) congener), all captured in extracellular-facing and drug-bound states. The structures revealed a multidrug-binding cavity festooned with four negatively charged amino acids and surprisingly limited hydrophobic moieties, in stark contrast to the general belief that aromatic amino acids play a prominent role in multidrug recognition. Furthermore, we discovered an uncommon cation-π interaction in the Na(+)-binding site located outside the drug-binding cavity and validated the biological relevance of both the substrate- and cation-binding sites by conducting drug resistance and transport assays. Additionally, we uncovered potential rearrangement of at least two transmembrane helices upon Na(+)-induced drug export. Based on our structural and functional analyses, we suggest that Na(+) triggers multidrug extrusion by inducing protein conformational changes rather than by directly competing for the substrate-binding amino acids. This scenario is distinct from the canonical antiport mechanism, in which both substrate and counterion compete for a shared binding site in the transporter. Collectively, our findings provide an important step toward a detailed and mechanistic understanding of multidrug transport.
- Department of Biochemistry and Molecular Biology, Rosalind Franklin University of Medicine and Science, North Chicago, IL 60064, USA. min.lu@rosalindfranklin.edu
Organizational Affiliation: