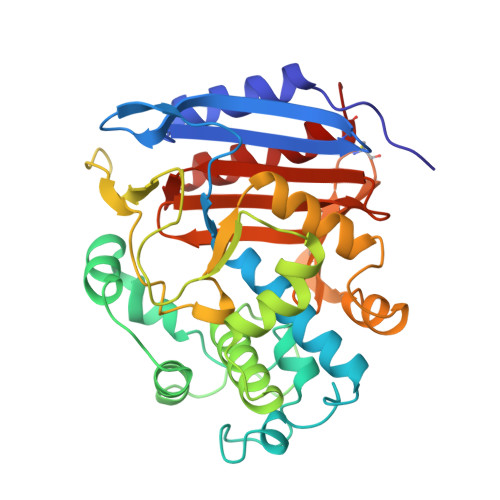
The refined crystallographic structure of a DD-peptidase penicillin-target enzyme at 1.6 A resolution.
Kelly, J.A., Kuzin, A.P.(1995) J Mol Biology 254: 223-236
- PubMed: 7490745 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.1995.0613
- Primary Citation Related Structures:
3PTE - PubMed Abstract:
The D-alanyl-D-alanine peptidase from Streptomyces sp. R61 is a 37,500 dalton exocellular enzyme that has served as a model for membrane-bound peptidases that are involved in bacterial cell wall biosynthesis. Inhibition of these enzymes by beta-lactam antibiotics ultimately leads to bacterial cell death. The X-ray crystal structure of the R61 D-alanyl-D-alanine peptidase has been solved using multiple isomorphous replacement, simulated annealing and least squares refinement. The space group and unit cell parameters are P2(1)2(1)2(1) with a = 51.1 A, b = 67.3 A and c = 102.4 A. The structure has been refined using 2 sigma data to 1.6 A resolution with a crystallographic R-factor of 0.148. The model contains 347 residues (2938 atoms) and 254 solvent molecules. The overall temperature factor is 9.6 A2, and the estimated coordinate error is 0.14 A. The protein consists of a single polypeptide chain organized into two regions. One region contains a nine-stranded antiparallel beta-sheet with helices on both faces; this region includes both the amino and carboxyl termini. The second region is all helical. Sixty percent of the residues occur in helices or beta-sheet. The reactive Ser62 is found between the two regions of the enzyme at the amino end of the protein's longest-helix which begins with one turn of 3(10) helix and continues with four turns of alpha-helix. The active site is an elongated pocket that contains four basic and four aromatic residues. An oxyanion hole is formed by Ser62 NH and Thr301 NH. The pocket also contains the few key residues that are conserved in all penicillin-binding proteins and beta-lactamases. Two of these residues, Lys65 and Tyr159, are among the 16 side-chains that take on multiple conformations in the R61 crystal structure. Three of the 12 proline rings adopt two conformations which we believe has not been previously reported. There is no anionic acid equivalent to the catalytic Glu166 found in Class A beta-lactamases. Two ordered water molecules (O507 and O644) are found buried in the active site and hydrogen-bonded to each other (2.6 A). O507 could potentially act as the hydrolytic water molecule for deacylation.
- Department of Molecular and Cell Biology, University of Connecticut, Storrs 06269-3125, USA.
Organizational Affiliation: