Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 A resolution. A comparison with the structure of glutathione reductase.
Mattevi, A., Schierbeek, A.J., Hol, W.G.(1991) J Mol Biol 220: 975-994
- PubMed: 1880807 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(91)90367-f
- Primary Citation Related Structures:
3LAD - PubMed Abstract:
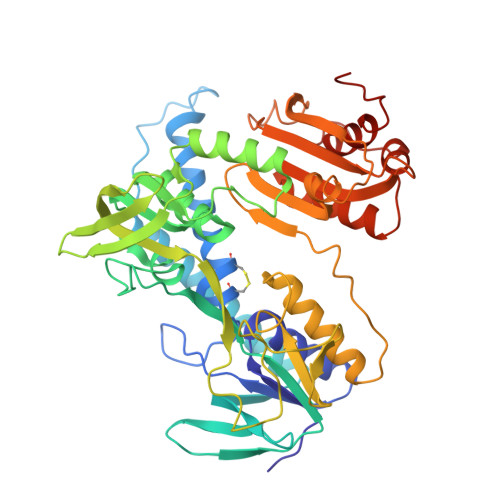
The structure of lipoamide dehydrogenase from Azotobacter vinelandii has been refined by the molecular dynamics technique to an R-factor of 19.8% at 2.2 A resolution. In the final model, the root-mean-square deviation from ideality is 0.02 A for bond lengths and 3.2 degrees for bond angles. The asymmetric unit comprises two subunits, each consisting of 466 amino acid residues and the prosthetic group FAD, plus 512 solvent molecules. The last ten amino acid residues of both chains are not visible in the electron density distribution and they are probably disordered. The operation required to superimpose the two chains forming the dimer is a rotation of exactly 180 degrees with no translation component. The final model shows the two independently refined subunits to be very similar, except for six loops located at the surface of the molecule. The structure of each subunit of the enzyme consists of four domains with the catalytic centre located at the subunit interface. The reactive disulphide bridge, 48-53, is oxidized with S gamma of Cys53 located 3.5 A away from carbon C-4a of the isoalloxazine ring. The side-chain of His450' points its N epsilon 2 towards S gamma of Cys48 and is hydrogen bonded to the carboxylate of Glu455'. The FAD is bound in an extended conformation and the isoalloxazine ring is not completely planar with an angle between the pteridine and the benzene ring of 7.3 degrees in the first subunit and of 12.1 degrees in the second one. The overall folding of lipoamide dehydrogenase is very similar to that of glutathione reductase. However, a comparison of the two enzymes, which have only 26% sequence identity, reveals significant conformational differences. These concern the tertiary as well as the quaternary structure of the two molecules. In each subunit of lipoamide dehydrogenase the NAD-binding domain and the interface domain appear to be differently oriented with respect to the FAD-binding domain by 7.1 degrees and 7.8 degrees, respectively. The interface domain contains, in addition, major changes in tertiary structure. Furthermore, the two subunits forming the dimer appear to be shifted with respect to each other by more than 4 A, when the lipoamide dehydrogenase dimer is compared with that of glutathione reductase. In spite of all these changes at the tertiary and quaternary level the active sites of the enzymes, which occur at the dimer interface, appear to be remarkably similar.(ABSTRACT TRUNCATED AT 400 WORDS)
- Department of Chemistry, University of Groningen, The Netherlands.
Organizational Affiliation: