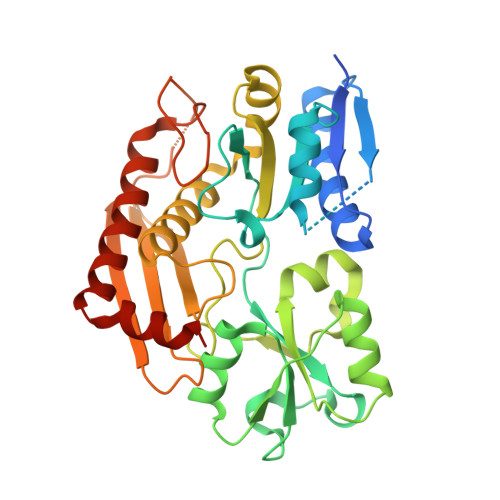
Structure of human porphobilinogen deaminase at 2.8 A: the molecular basis of acute intermittent porphyria
Gill, R., Kolstoe, S.E., Mohammed, F., Al D-Bass, A., Mosely, J.E., Sarwar, M., Cooper, J.B., Wood, S.P., Shoolingin-Jordan, P.M.(2009) Biochem J 420: 17-25
- PubMed: 19207107 Search on PubMed
- DOI: https://doi.org/10.1042/BJ20082077
- Primary Citation Related Structures:
3EQ1 - PubMed Abstract:
Mutations in the human PBGD (porphobilinogen deaminase) gene cause the inherited defect AIP (acute intermittent porphyria). In the present study we report the structure of the human uPBGD (ubiquitous PBGD) mutant, R167Q, that has been determined by X-ray crystallography and refined to 2.8 A (1 A=0.1 nm) resolution (Rfactor=0.26, Rfree=0.29). The protein crystallized in space group P2(1)2(1)2 with two molecules in the asymmetric unit (a=81.0 A, b=104.4 A and c=109.7 A). Phases were obtained by molecular replacement using the Escherichia coli PBGD structure as a search model. The human enzyme is composed of three domains each of approx. 110 amino acids and possesses a dipyrromethane cofactor at the active site, which is located between domains 1 and 2. An ordered sulfate ion is hydrogen-bonded to Arg26 and Ser28 at the proposed substrate-binding site in domain 1. An insert of 29 amino acid residues, present only in mammalian PBGD enzymes, has been modelled into domain 3 where it extends helix alpha2(3) and forms a beta-hairpin structure that contributes to a continuous hydrogen-bonding network spanning domains 1 and 3. The structural and functional implications of the R167Q mutation and other mutations that result in AIP are discussed.
- Centre for Amyloidosis and Acute Phase Proteins, Department of Medicine, UCL Medical School, Rowland Hill Street, London NW3 2PF, UK.
Organizational Affiliation: