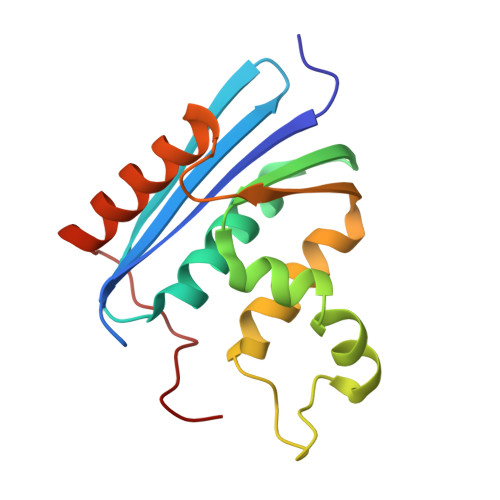
Structural details of ribonuclease H from Escherichia coli as refined to an atomic resolution.
Katayanagi, K., Miyagawa, M., Matsushima, M., Ishikawa, M., Kanaya, S., Nakamura, H., Ikehara, M., Matsuzaki, T., Morikawa, K.(1992) J Mol Biol 223: 1029-1052
- PubMed: 1311386 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(92)90260-q
- Primary Citation Related Structures:
2RN2 - PubMed Abstract:
The crystal structure of RNase H from Escherichia coli has been determined by the multiple isomorphous replacement method, and refined by the stereochemically restrained least-squares procedure to a crystallographic R-factor of 0.196 at 1.48 A resolution. In the final structure, the root-mean-square (r.m.s.) deviation for bond lengths is 0.017 A, and for angle distances 0.036 A. The structure is composed of a five-stranded beta-sheet and five alpha-helices, and reveals the details of hydrogen bonding, electrostatic and hydrophobic interactions between intra- and intermolecular residues. The refined structure allows an explanation of the particular interactions between the basic protrusion, consisting of helix alpha III and the following loop, and the remaining major domain. The beta-sheet, alpha II, alpha III and alpha IV form a central hydrophobic cleft that contains all six tryptophan residues, and presumably serves to fix the orientation of the basic protrusion. Two parallel adjacent helices, alpha I and alpha IV, are associated with a few triads of hydrophobic interactions, including many leucine residues, that are similar to the repeated leucine motif. The well-defined electron density map allows detailed discussion of amino acid residues likely to be involved in binding a DNA/RNA hybrid, and construction of a putative model of the enzyme complexed with a DNA/RNA hybrid oligomer. In this model, a protein region, from the Mg(2+)-binding site to the basic protrusion, covers roughly two turns of a DNA/RNA hybrid double helix. A segment (11-23) containing six glycine residues forms a long loop between the beta A and beta B strands. This loop, which protrudes into the solvent region, lies on the interface between the enzyme and a DNA/RNA hybrid in the model of the complex. The mean temperature factors of main-chain atoms show remarkably high values in helix alpha III that constitutes the basic protrusion, suggesting some correlation between its flexibility and the nucleic acid binding function. The Mg(2+)-binding site, surrounded by four invariant acidic residues, can now be described more precisely in conjunction with the catalytic activity. The arrangement of molecules within the crystal appears to be dominated by the cancelling out of a remarkably biased charge distribution on the molecular surface, which is derived in particular from the separation between the acidic Mg(2+)-binding site and the basic protrusion.
- Protein Engineering Research Institute, Osaka, Japan.
Organizational Affiliation: