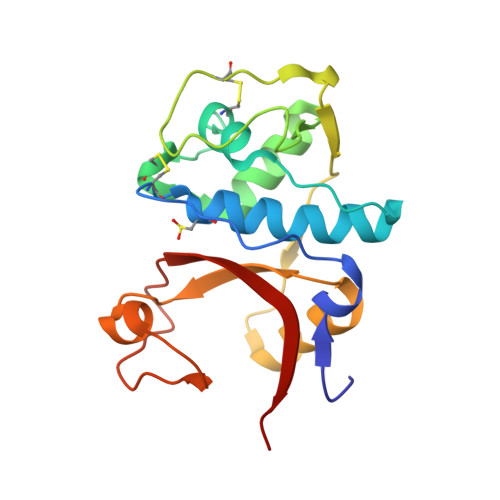
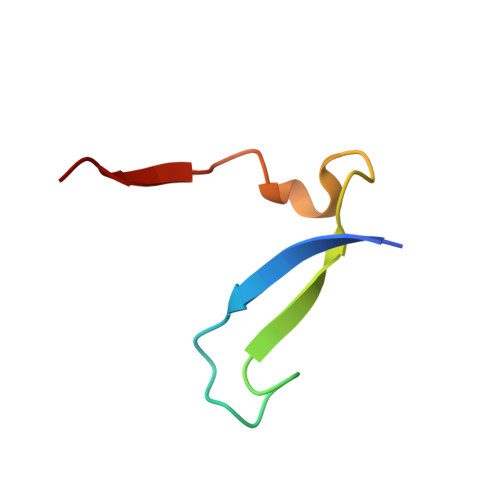
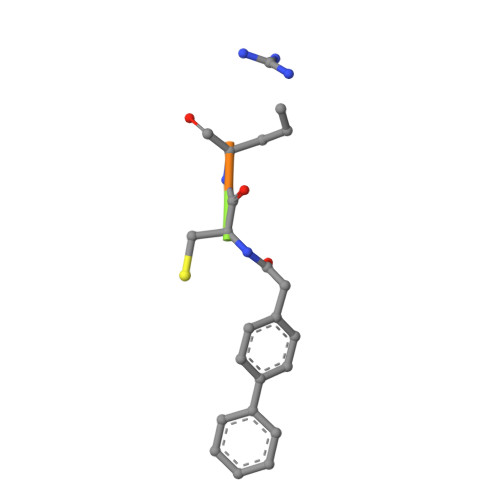
Design of non-covalent inhibitors of human cathepsin L. From the 96-residue proregion to optimized tripeptides
Chowdhury, S., Sivaraman, J., Wang, J., Devanathan, G., Lachance, P., Qi, H., Menard, R., Lefebvre, J., Konishi, Y., Cygler, M., Sulea, T., Purisima, E.O.(2002) J Med Chem 45: 5321-5329
- PubMed: 12431059 Search on PubMed
- DOI: https://doi.org/10.1021/jm020238t
- Primary Citation Related Structures:
1MHW - PubMed Abstract:
A novel series of noncovalent inhibitors of cathepsin L have been designed to mimic the mode of autoinhibition of procathepsin L. Just like the propeptide, these peptide-based inhibitors have a reverse-binding mode relative to a substrate and span both the S' and S subsites of the enzyme active site. In contrast to previous studies in which even moderate truncation of the full-length propeptide led to rapid reduction in potency, these blocked tripeptide-sized inhibitors maintain nanomolar potency. Moreover, these short peptides show higher selectivity (up to 310-fold) for inhibiting cathepsin L over K versus only 2-fold selectivity of the 96-residue propeptide of cathepsin L. A 1.9 A X-ray crystallographic structure of the complex of cathepsin L with one of the inhibitors confirms the designed reverse-binding mode of the inhibitor as well as its noncovalent nature. Enzymatic analysis also shows the inhibitors to be resistant to hydrolysis at elevated concentrations of the enzyme. The mode of inhibition of these molecules provides a general strategy for inhibiting other cathepsins as well as other proteases.
- Biotechnology Research Institute, National Research Council of Canada, 6100 Royalmount Avenue, Montreal, Quebec, H4P 2R2, Canada.
Organizational Affiliation: