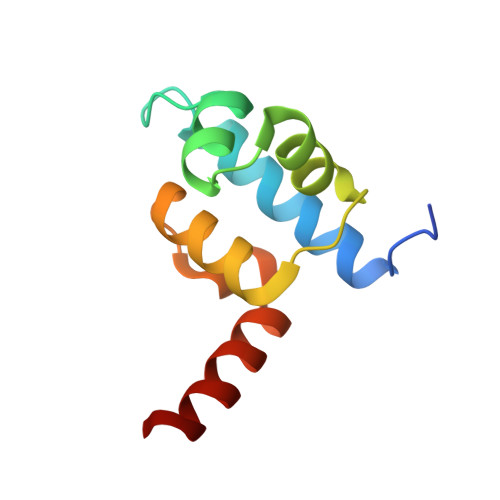
The crystal structure of a mutant protein with altered but improved hydrophobic core packing.
Lim, W.A., Hodel, A., Sauer, R.T., Richards, F.M.(1994) Proc Natl Acad Sci U S A 91: 423-427
- PubMed: 8278404 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.91.1.423
- Primary Citation Related Structures:
1LLI - PubMed Abstract:
The dense packing observed in protein interiors appears to be crucial for stabilizing the native structure--even subtle internal substitutions are usually destabilizing. Thus, steric complementarity of core residues is thought to be an important criterion for "inverse folding" predictive methods, which judge whether a newly determined sequence is consistent with any known folds. A major problem in the development of useful core packing evaluation algorithms, however, is that there are occasional mutations that are predicted to disrupt native packing but that yield an equally or more stable protein. We have solved the crystal structure of such a variant of lambda repressor, which, despite having three larger core substitutions, is more stable than the wild type. The structure reveals that the protein accommodates the potentially disruptive residues with shifts in its alpha-helical arrangement. The variant is apparently more stable because its packing is improved--the core has a higher packing density and little geometric strain. These rearrangements, however, cause repositioning of functional residues, which result in reduced DNA binding activity. By comparing these results with the predictions of two core packing algorithms, it is clear that the protein possesses a relatively high degree of main-chain flexibility that must be accounted for in order to predict the full spectrum of compatible core sequences. This study also shows how, in protein evolution, a particular set of core residue identities might be selected not because they provide optimal stability but because they provide sufficient stability in addition to the precise structure required for optimal activity.
- Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511.
Organizational Affiliation: