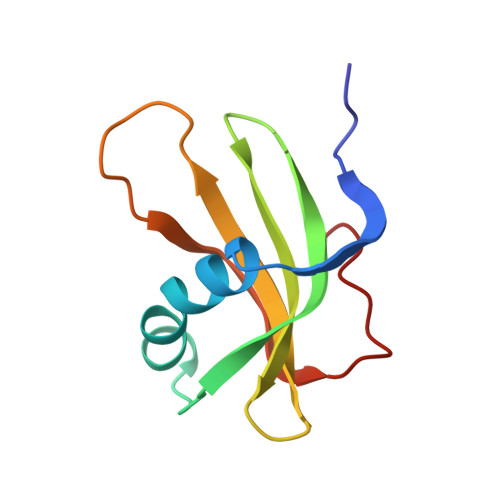
The three-dimensional solution structure of human stefin A.
Martin, J.R., Craven, C.J., Jerala, R., Kroon-Zitko, L., Zerovnik, E., Turk, V., Waltho, J.P.(1995) J Mol Biology 246: 331-343
- PubMed: 7869384 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.1994.0088
- Primary Citation Related Structures:
1DVC, 1DVD - PubMed Abstract:
The three-dimensional solution structure of recombinant human stefin A has been determined by a simulated annealing protocol using a total of 1113 distance and angle constraints obtained from 1H and 15N HMR spectroscopy. The solution structure is represented by a family of 17 conformers with an average root-mean-square deviation relative to the mean structure of 0.44 A for backbone atoms and 0.94 A for all heavy atoms for the main body of the structure. The protein has a well-defined global fold consisting of five anti-parallel beta-strands wrapped around a central five-turn alpha-helix. There is considerable similarity between the structural features of free stefin A in solution and the X-ray structure of the homologous protein stefin B in its complex with papain, but there are also some important differences in the regions which are fundamental to proteinase binding. The differences consist primarily of two regions of high conformational heterogeneity in free stefin A which correspond in stefin B to two of the components of the tripartite wedge that docks into the active site of the target proteinase. These regions, which are shown to be mobile in solution, are the five N-terminal residues and the second binding loop. In the bound conformation of stefin B they form a turn and a short helix, respectively.
- Krebs Institute, Department of Molecular Biology and Biotechnology, University of Sheffield, U.K.
Organizational Affiliation: