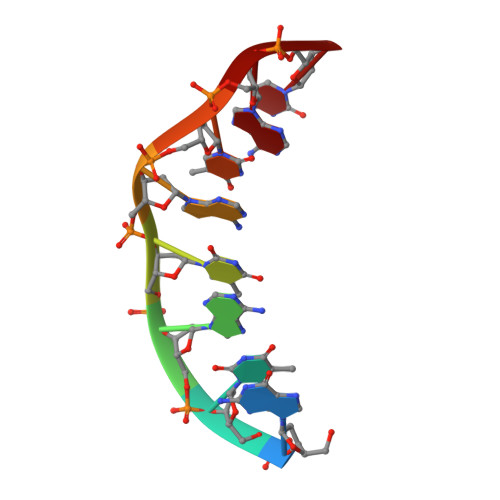
Solution structure of [d(GTATATAC)]2 via restrained molecular dynamics simulations with nuclear magnetic resonance constraints derived from relaxation matrix analysis of two-dimensional nuclear Overhauser effect experiments.
Schmitz, U., Pearlman, D.A., James, T.L.(1991) J Mol Biology 221: 271-292
- PubMed: 1920410 Search on PubMed
- DOI: https://doi.org/10.1016/0022-2836(91)80219-k
- Primary Citation Related Structures:
1D42 - PubMed Abstract:
Two-dimensional nuclear Overhauser effect (2D NOE) spectra have been used as the experimental basis for determining the solution structure of the duplex [d(GTATATAC)]2 employing restrained molecular dynamics (rMD) simulations. The MARDIGRAS algorithm has been employed to construct a set of 233 interproton distance constraints via iterative complete relaxation matrix analysis utilizing the peak intensities from the 2D NOE spectra obtained for different mixing times and model structures. The upper and lower bounds for each of the constraints, defining size of a flat-well potential function term used in the rMD simulations, were conservatively chosen as the largest or smallest value calculated by MARDIGRAS. Three different starting models were utilized in several rMD calculations: energy-minimized A-DNA, B-DNA, and a structure containing wrinkled D-DNA in the interior. Considerable effort was made to define the appropriate force constants to be employed with the NOE terms in the AMBER force field, using as criteria the average constraints deviation, the constraints violation energy and the total energy. Of the 233 constraints, one was generated indirectly, but proved to be crucial in defining the structure: the cross-strand A5-H2 A5-H2 distance. As those two protons resonate isochronously for the self-complementary duplex, the distance cannot be determined directly. However, the general pattern of 2D NOE peak intensities, spin-lattice relaxation time (T1) values, and 31P nuclear magnetic resonance spectra lead to use of the A3-H2 A7-H2 distance for A5-H2 A5-H2 as well. Five rMD runs, with different random number seeds, were made for each of the three starting structures with the full distance constraint set. The average structure from all 15 runs and the five-structure averages from each starting structure were all quite similar. Two rMD runs for each starting structure were made with the A5-H2 A5-H2 constraint missing. The average of these six rMD runs revealed differences in structure, compared to that with the full set of constraints, primarily for the middle two base-pairs involving the missing cross-strand constraint but global deviations also were found. Conformational analysis of the resulting structures revealed that the inner four to six base-pairs differed in structure from the termini. Furthermore, an alternating structure was suggested with features alternating for the A-T and T-A steps.
- Department of Pharmaceutical Chemistry, University of California, San Francisco 94143-0446.
Organizational Affiliation: