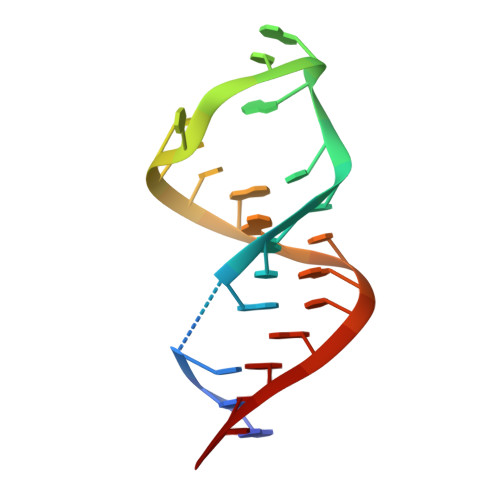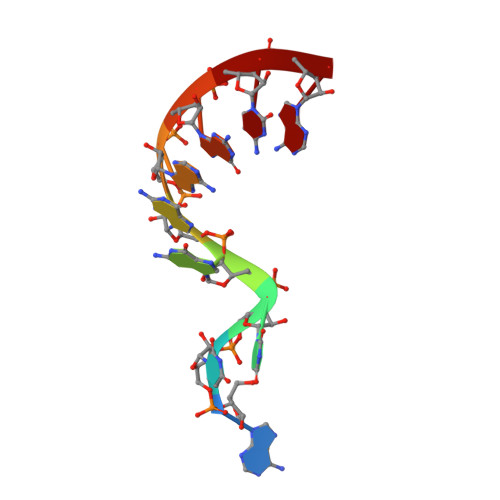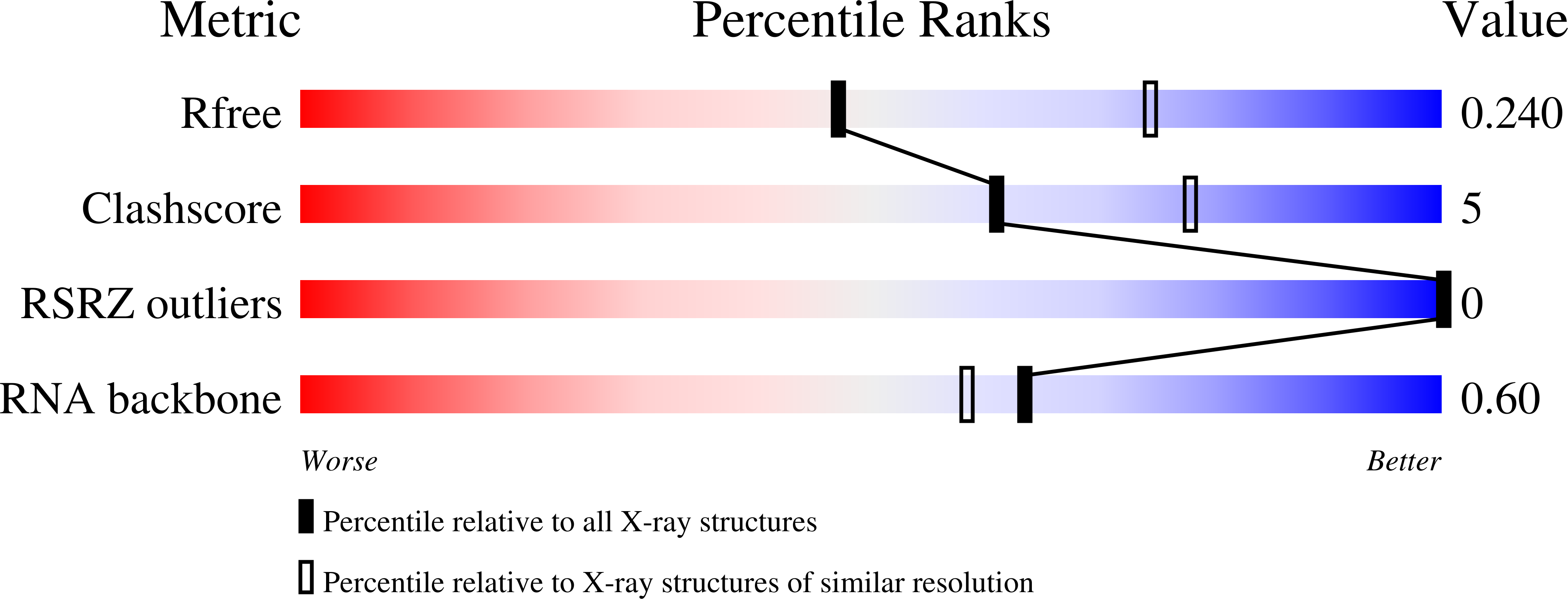
Crystal structure and ligand-induced folding of the SAM/SAH riboswitch.
Huang, L., Liao, T.W., Wang, J., Ha, T., Lilley, D.M.J.(2020) Nucleic Acids Res 48: 7545-7556
- PubMed: 32520325
- DOI: https://doi.org/10.1093/nar/gkaa493
- Primary Citation of Related Structures:
6YL5, 6YLB, 6YMI, 6YMJ, 6YMK, 6YML, 6YMM - PubMed Abstract:
While most SAM riboswitches strongly discriminate between SAM and SAH, the SAM/SAH riboswitch responds to both ligands with similar apparent affinities. We have determined crystal structures of the SAM/SAH riboswitch bound to SAH, SAM and other variant ligands at high resolution. The riboswitch forms an H-type pseudoknot structure with coaxial alignment of the stem-loop helix (P1) and the pseudoknot helix (PK). An additional three base pairs form at the non-open end of P1, and the ligand is bound at the interface between the P1 extension and the PK helix. The adenine nucleobase is stacked into the helix and forms a trans Hoogsteen-Watson-Crick base pair with a uridine, thus becoming an integral part of the helical structure. The majority of the specific interactions are formed with the adenosine. The methionine or homocysteine chain lies in the groove making a single hydrogen bond, and there is no discrimination between the sulfonium of SAM or the thioether of SAH. Single-molecule FRET analysis reveals that the riboswitch exists in two distinct conformations, and that addition of SAM or SAH shifts the population into a stable state that likely corresponds to the form observed in the crystal. A model for translational regulation is presented whereby in the absence of ligand the riboswitch is largely unfolded, lacking the PK helix so that translation can be initiated at the ribosome binding site. But the presence of ligand stabilizes the folded conformation that includes the PK helix, so occluding the ribosome binding site and thus preventing the initiation of translation.
Organizational Affiliation:
Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, P. R. China.