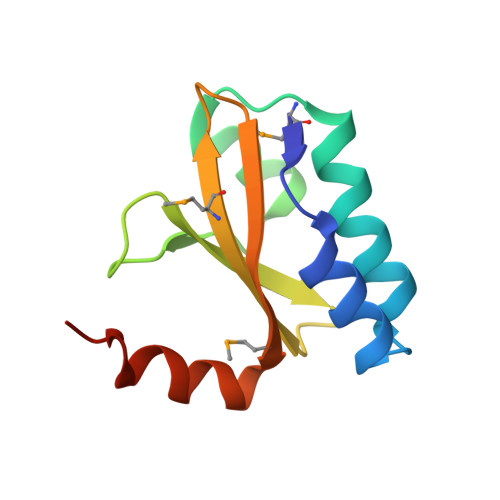
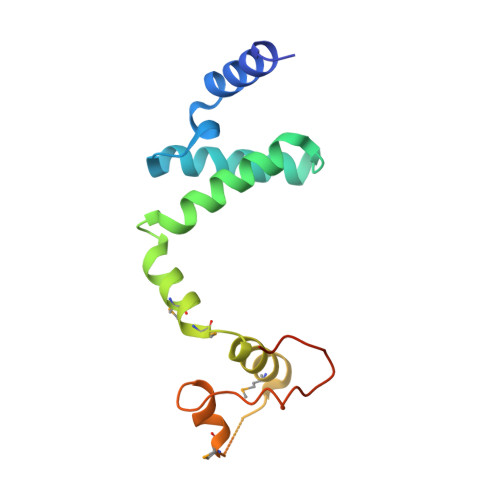
Structural insight into the E. coli HigBA complex
Yang, J., Zhou, K., Liu, P., Dong, Y., Gao, Z., Zhang, J., Liu, Q.(2016) Biochem Biophys Res Commun 478: 1521-1527
- PubMed: 27601326
- DOI: https://doi.org/10.1016/j.bbrc.2016.08.131
- Primary Citation of Related Structures:
5IFG - PubMed Abstract:
The toxin-antitoxin system is ubiquitously existed in bacteria and archaea, performing a wide variety of functions modulating cell fitness in response to environmental cues. In this report, we solved the crystal structure of the toxin-antitoxin HigBA complex from E. coli K-12 to 2.7 Å resolution. The crystal structure of the HigBA complex displays a hetero-tetramer (HigBA)2 form comprised by two HigB and two HigA subunits. Each toxin HigB resumes a microbial RNase T1 fold, characteristic of a three antiparallel β-sheet core shielded by a few α-helices at either side. Each antitoxin HigA composed of all α-helices resembles a "C"-shaped clamp nicely encompassing a HigB in the (HigBA)2 complex. Two HigA monomers dimerize at their N-terminal domain. We showed that HigA helix α1 was essential for HigA dimerization and the hetero-tetramer (HigBA)2 formation, but not for a hetero-dimeric HigBA formation. HigA dimerization mediated by helix α1 was dispensable for DNA-binding, as a heterodimeric HigBA complex still bound to the higBA operator in vitro. The HigA C-terminal domain with a helix-turn-helix fold was essential for DNA binding. We also defined two palindromes in higBA operator specifically recognized by HigA and HigBA in vitro.
Organizational Affiliation:
School of Life Sciences, University of Dalian Science and Technology, Dalian, Liaolin Province, 230027, People's Republic of China.