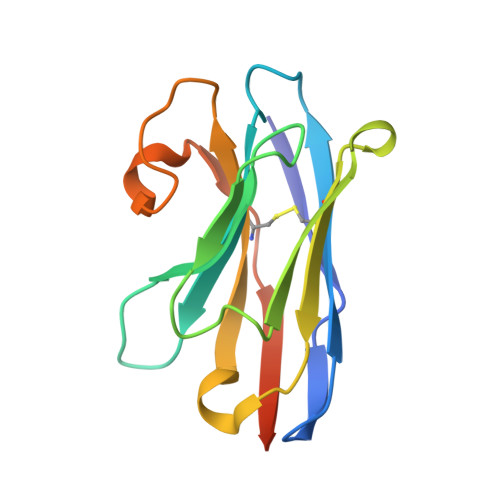
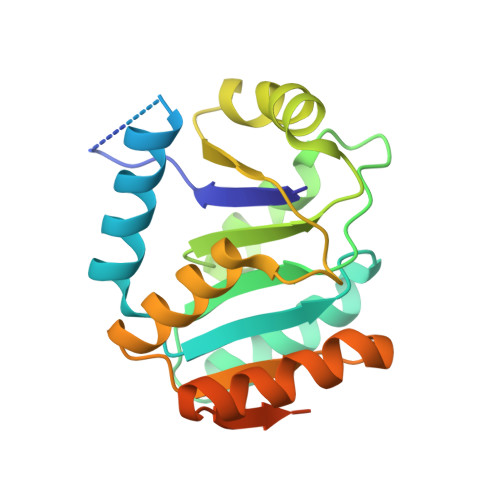
The unexpected structure of the designed protein Octarellin V.1 forms a challenge for protein structure prediction tools.
Figueroa, M., Sleutel, M., Vandevenne, M., Parvizi, G., Attout, S., Jacquin, O., Vandenameele, J., Fischer, A.W., Damblon, C., Goormaghtigh, E., Valerio-Lepiniec, M., Urvoas, A., Durand, D., Pardon, E., Steyaert, J., Minard, P., Maes, D., Meiler, J., Matagne, A., Martial, J.A., Van de Weerdt, C.(2016) J Struct Biol 195: 19-30
- PubMed: 27181418
- DOI: https://doi.org/10.1016/j.jsb.2016.05.004
- Primary Citation of Related Structures:
4ZV6, 5BOP - PubMed Abstract:
Despite impressive successes in protein design, designing a well-folded protein of more 100 amino acids de novo remains a formidable challenge. Exploiting the promising biophysical features of the artificial protein Octarellin V, we improved this protein by directed evolution, thus creating a more stable and soluble protein: Octarellin V.1. Next, we obtained crystals of Octarellin V.1 in complex with crystallization chaperons and determined the tertiary structure. The experimental structure of Octarellin V.1 differs from its in silico design: the (αβα) sandwich architecture bears some resemblance to a Rossman-like fold instead of the intended TIM-barrel fold. This surprising result gave us a unique and attractive opportunity to test the state of the art in protein structure prediction, using this artificial protein free of any natural selection. We tested 13 automated webservers for protein structure prediction and found none of them to predict the actual structure. More than 50% of them predicted a TIM-barrel fold, i.e. the structure we set out to design more than 10years ago. In addition, local software runs that are human operated can sample a structure similar to the experimental one but fail in selecting it, suggesting that the scoring and ranking functions should be improved. We propose that artificial proteins could be used as tools to test the accuracy of protein structure prediction algorithms, because their lack of evolutionary pressure and unique sequences features.
Organizational Affiliation:
GIGA-Research, Molecular Biomimetics and Protein Engineering, University of Liège, Liège, Belgium. Electronic address: maxifigueroa@udec.cl.