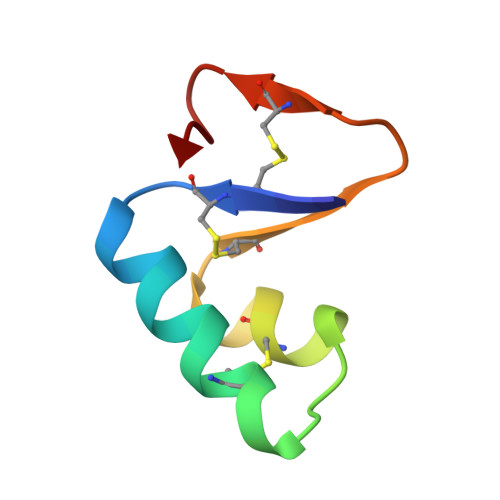
Atomic resolution (0.83 A) crystal structure of the hydrophobic protein crambin at 130 K.
Teeter, M.M., Roe, S.M., Heo, N.H.(1993) J Mol Biol 230: 292-311
- PubMed: 8450543
- DOI: https://doi.org/10.1006/jmbi.1993.1143
- Primary Citation of Related Structures:
1CBN - PubMed Abstract:
To enhance the already high quality of diffraction data for crystals of the hydrophobic protein crambin, X-ray data were collected at 130 K by the method of H. Hope to 0.83 A resolution. Refinement with PROLSQ yields a model with an R value of 10.5%. The final model had three parameter anisotropic vibration factors for all atoms, which included 367 protein heavy atoms, 372 hydrogen atoms and 144 solvent atoms with one ethanol molecule. Dihedral angles and hydrogen-bonding distances generally agree with earlier studies of high-resolution protein structures, but some new patterns are noted. Solvent-related helix distortions are reminiscent of those described by others. Helix and beta-sheet regions show distinct patterns in their side-chain conformations. Despite crambin's hydrophobic nature, its accessible surface area in the crystal is surprisingly close to that of water-soluble proteins like myoglobin and carboxypeptidase A. More of crambin's hydrophobic surface is buried in the crystal, perhaps accounting for its high order of diffraction. A total of 24% of the 46 residues show discrete disorder at 130 K. This includes five side-chains at both 300 and 130 K, and six more side-chains and an ethanol molecule at 130 K. Disorder is associated with the sequence microheterogeneity at Pro/Ser22 and Leu/Ile25, with space filling or with solvent disorder. Correlated conformations extend over three to five residues. The patterns of disorder in this structure reveal important principles of protein structure and its dynamics. Finding disordered groups correlated over 5 to 8 A suggests that co-ordinated motion extends in groups rather than simply as uncorrelated movement around an atom center. Thermal diffuse scattering experiments on insulin and lysozyme are consistent with this interpretation. Nearly all of the protein-bound solvent has been located. Less than 1% of protein accessible surface area remains uncovered by solvent or crystal contacts. Preliminary analysis of the solvent network reveals two main networks in each of four solvent regions.
Organizational Affiliation:
Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02167.